Поступило в редакцию: Декабрь 2025
УДК 661.48
Fluorine Notes, 2026, 164, 1-2
НЕКОТОРЫЕ НАПРАВЛЕНИЯ ХИМИИ ФТОРА. СБОРНИК СООБЩЕНИЙ
СООБЩЕНИЕ 2. ЧАСТЬ 3. ФТОРИРОВАНИЕ АЛИФАТИЧЕСКИХ СОЕДИНЕНИЙ ВЫСШИМИ ФТОРИДАМИ МЕТАЛЛОВ ПЕРЕМЕННОЙ ВАЛЕНТНОСТИ В РАБОТАХ РОССИЙСКИХ ИССЛЕДОВАТЕЛЕЙ
В.В. Корнилов
Аннотация: Приведены результаты фторирования метана, ацетилена, этилена и ряда полифорэтанов высшими фторидами металлов переменной валентности (HVMFs). Рассмотрены возможные закономерности взаимодействия С1-С2 углеводородов и С1-С2 полифторалканов с HVMFs. Рассмотрены вероятные механизмы фторирования и перегруппировок, связанных с H-миграцией, на промежуточных стадиях фторирования. Рассмотрено влияние на соотношение изомеров полифторэтанов адсорбции молекул реагентов на поверхности HVMFs.
Ключевые слова: фториды металлов переменной валентности, трифторид кобальта, трифторид марганца, тетрафторид церия, изомеры тетрафторэтана, карбкатион.
Введение
В предыдущих частях сообщения [1,2] был представлен краткий обзор по фторированию органических соединений высшими фторидами металлов переменной валентности (HVMFs), рассмотрены вероятные механизмы фторирования ароматических и алифатических соединений с точки зрения катион-радикальной теории на основе публикаций зарубежных авторов. В третьей части сообщения сделан обзор работ отечественных исследователей в области фторирования алифатических соединений высшими фторидами металлов переменной валентности.
2. Фторирование С1-С2 углеводородов и С1-С2 полифторалканов высшими фторидами металлов переменной валентности
Взаимодействие низших углеводородов (метана, этилена и др.) и их фторпроизводных с высшими фторидами кобальта, марганца и церия изучалось в 70х-90х годах XX века в РНЦ «Прикладная химия» (до 1992 г. – ГИПХ) (Санкт-Петербург). Значительная часть результатов была опубликована в работах [3-9] и обзорной статье [10].
Для изучения процессов применяли вертикальные реактора с внутренним диаметром 18-36 мм и высотой 45-100 см, исходные соединения подавали в нижнюю часть реактора. Такая конструкция обладала рядом преимуществ для исследований по сравнению с горизонтальными реакторами с перемешивающими устройствами, которые использовались зарубежными авторами [1,2].
Во-первых, вертикальные реактора имели простую конструкцию и, соответственно, повышенную надежность.
Во-вторых, горизонтальные реактора с мешалками заполнялись фторидами металлов на ≈2/3÷5/6 своего объема, поэтому имели в верхней части реактора пустое пространство. Вертикальные реактора заполнялись фторирующим реагентом полностью, что обеспечивало равномерное прохождение газообразного углеводорода через весь слой HVMFs и исключало проскок углеводорода без взаимодействия с высшим фторидом даже при малых временах контакта (несколько секунд). Экспериментально было установлено, что мелкодисперсное состояние и равномерность засыпки сохранялись неизменными даже после большого количества циклов регенерация – синтез.
В-третьих, применяемая конструкция позволяла минимизировать погрешность при измерениях температурного профиля реактора. Для этого реактора были снабжены либо несколькими тонкостенными карманами для термопар (через 10-12 см по высоте), либо тонкостенной трубкой (d=3 мм), проходящей вдоль оси реактора. В последнем случае профиль температур получали перемещением термопары внутри трубки.
В некоторых исследованиях для изучения зависимости состава продуктов от величины слоя HVMFs использовали реактор, который был снабжен несколькими пробоотборниками по высоте.
Было установлено, что при фторировании метана, этилена, ацетилена и др. без разбавления инертным газом реакция проходила в узкой зоне слоя фторидов (2-5 см) с постепенным смещением зоны реакции по высоте реактора. Смещение такой зоны можно было контролировать по профилю температуры. При приближении зоны взаимодействия наблюдался резкий рост температуры с прохождением значения через пик и последующим снижением. Величина температурного градиента зависела от скорости подачи углеводорода и возможностью отвода тепла в реакторе. Если значение пика температуры достигало ≈250ºС, то для фторидов кобальта наблюдали практически полное превращение трифторида кобальта в дифторид. Это было установлено следующим образом: процесс останавливали при нахождении температурного пика примерно в средней части реактора, реактор продували нейтральным газом, охлаждали и разбирали. Прореагировавший слой состоял из дифторида кобальта пурпурного цвета. Непрореагировавший слой выше зоны реакции представлял из себя практически чистый трифторид кобальта коричневого цвета.
Применение разбавления исходного углеводорода инертным газом позволяло проводить исследования в изотермическом режиме. В этом случае рабочей зоной был весь слой HVMFs.
2.1. Фторирование метана трифторидом кобальта
В работе [3] были опубликованы результаты фторирования метана, дифторметана и трифторметана трифторидом кобальта (Рис.1). В статье [8] приведены более подробные данные по фторированию дифторметана в диапазоне температур 184÷351ºC и трифторметана в диапазоне температур 210÷362ºC.
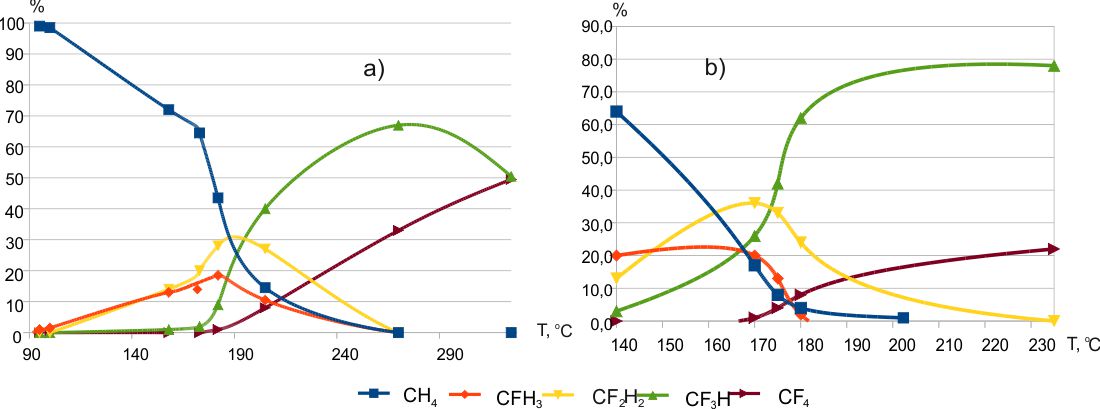
Рис. 1. Зависимость состава продуктов фторирования метана трифторидом кобальта от температуры (а- 5% смесь CH4 и гелия, b- чистый CH4) [3].
По аналогии с фторированием других углеводородов было предложено, что процесс протекает по катион-радикальному механизму (Схема 1).

Схема 1.
Очевидно, что фторирование метана не может протекать через образование промежуточных алкенов. Поскольку не просматриваются принципиальные отличия процесса фторирования метана от других углеводородов, можно предположить, что тезис об образовании олефинов на первоначальном этапе фторирования алканов (например, пропана [11]) представляется небесспорным (см. предыдущую часть сообщения [2]).
2.2. Фторирование ацетилена высшими фторидами кобальта и церия
В работах [4,5] были представлены результаты фторирования ацетилена высшими фторидами церия и кобальта.
Было установлено, что фторирование ацетилена тетрафторидом церия начиналось при температуре выше 100ºС (Рис.2). Тетрафторэтаны (на рисунке представлено суммарное содержание 1,1,2,2- и 1,1,1,2-тетрафторэтанов в соотношении ≈4:1) и пентафторэтан образуются в достаточно широком интервале температур.
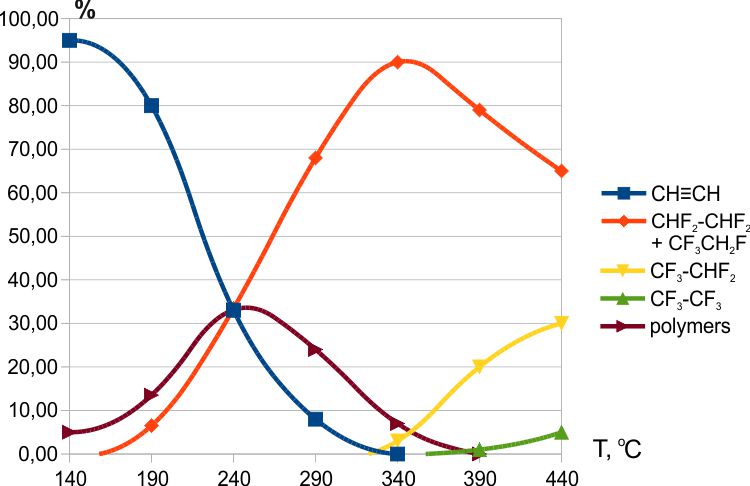
Рис. 2. Зависимость состава продуктов фторирования ацетилена тетрафторидом церия от температуры.
Особенностью взаимодействия ацетилена с тетрафторидом церия являлось наличие в продуктах взаимодействия жидких при нормальных условиях олигомерных соединений. Возможный механизм образования таких соединений будет рассмотрен ниже (п. 2.4.4).
Взаимодействие ацетилена с CoF3 начиналось при ≈ 80°С, при 250°С в продуктах взаимодействия образовывалось более 50% пентафторэтана (Рис.3) [5].
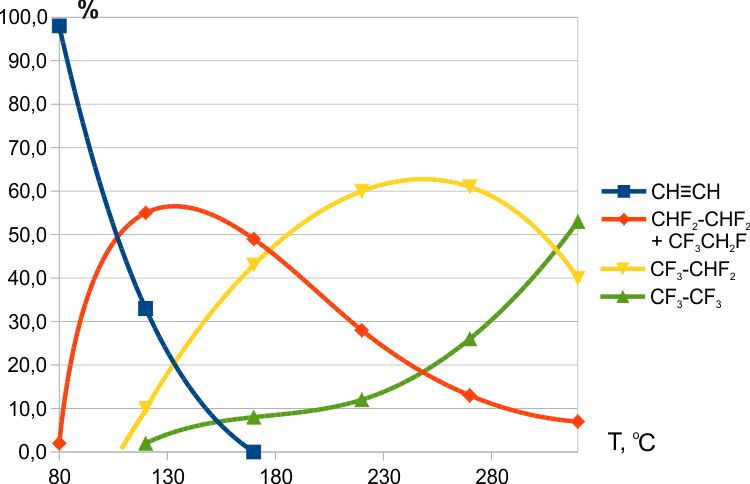
Рис. 3. Зависимость состава продуктов фторирования ацетилена трифторидом кобальта от температуры.
На Рис.3 представлено суммарное содержание двух изомеров тетрафторэтана с соотношением 1,1,2,2-тетрафторэтана к 1,1,1,2-тетрафторэтану как ≈3:1. Возможный механизм появления 1,1,1,2-тетрафторэтана также будет рассмотрен ниже (см. п. 2.4.3).
2.3. Фторирование этилена высшими фторидами кобальта, марганца и церия
В работах [4-6] были представлены результаты фторирования этилена и некоторых полифторалканов высшими фторидами кобальта, марганца и церия (Рис. 4-6). Было установлено, что в случае трифторида кобальта реакция с этиленом начиналась уже при комнатной температуре, при 50ºС следы этилена в продуктах реакции отсутствовали. При температурах выше 80-100ºС в продуктах могли присутствовать незначительные количества полифторметанов. Реакция этилена с трифторидом марганца начиналась при ≈140 ºС, присутствие этилена сохранялось до 260÷270 ºС. Реакция этилена с тетрафторидом церия начиналась при ≈125 ºС, следы этилена сохранялись до 235ºС. Кроме указанных на Рис. 6 соединений в продуктах реакции при низких температурах в следовых количествах был обнаружен фторэтилен.
Полученные результаты подтверждали тезис зависимости силы фторирующего реагента от окислительного потенциала катиона металла. Начало взаимодействия тетрафторида церия с этиленом при более низкой температуре по сравнению с более сильным трифторидом марганца можно объяснить протеканием процесса через конкурирующий механизм ионной (радикальной) полимеризации, который приводил к образованию жидких полимерных продуктов (см. п. 2.4.4).
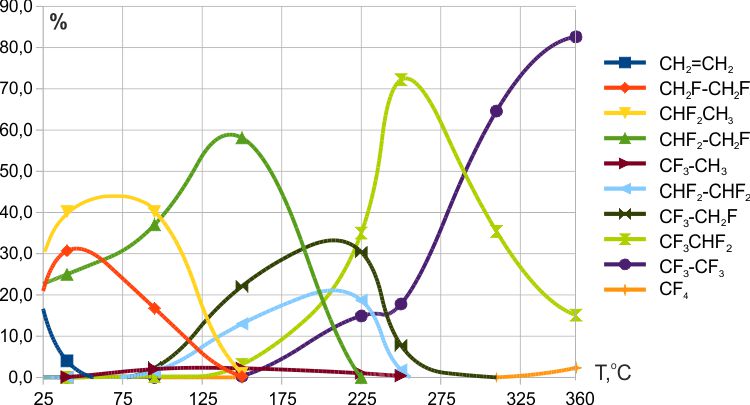
Рис. 4. Зависимость состава продуктов фторирования этилена трифторидом кобальта от температуры (интервал 25→310ºС, время контакта ≈7 мин.).
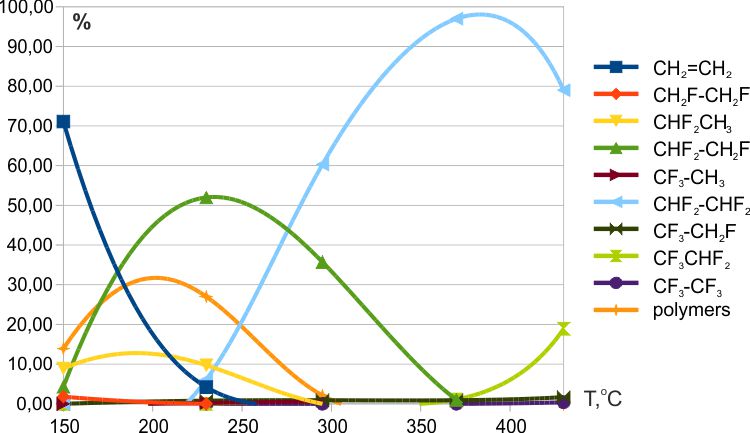
Рис. 5. Зависимость состава продуктов фторирования этилена тетрафторидом церия от температуры (интервал 150→430ºС, время контакта ≈7 мин.).
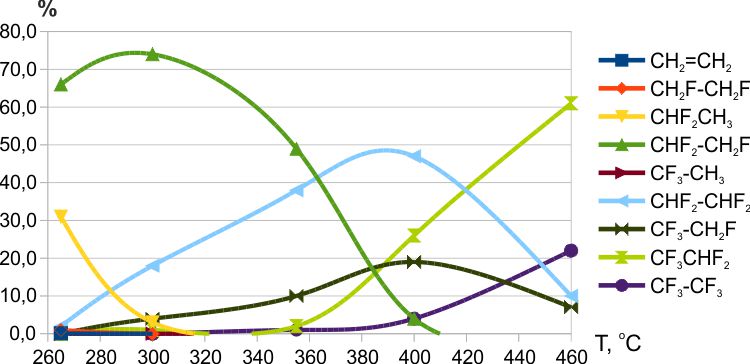
Рис. 6. Зависимость состава продуктов фторирования этилена трифторидом марганца от температуры (интервал 265-460ºС, время контакта ≈7 мин.).
2.4. Возможные закономерности взаимодействия С1-С2 углеводородов и С1-С2 полифторалканов с HVMFs
На основании полученных результатов было сделано несколько выводов и предположений, которые могут улучшить представления о процессах взаимодействия алифатических соединений с HVMFs.
2.4.1. Вероятный механизм образования 1,1-дифторэтана
Была подтверждена возможность перегруппировки карбкатиона (I) с образованием 1,1-дифторэтана (Схема 2).

Схема 2. Возможный механизм образования 1,1-дифторэтана при фторировании CoF3.
Впервые предположение о такой перегруппировке было сделано Д. Бурдоном с коллегами в 1976 г. [12] на основании данных Д. Кларка по стабильности карбкатионов I и II (см. [2]). В сообщении [5] было показано, что при фторировании этилена трифторидом кобальта 1,1-дифторэтан присутствовал в продуктах реакции в значительной концентрации уже при 40 ºC. С другой стороны, сам 1,2-дифторэтан не взаимодействовал с трифторидом кобальта температурах ниже 50 ºC, то есть изомеризации 1,2-дифторэтана в 1,1-дифторэтан при 40 ºC исключалась. Поэтому вывод о появлении 1,1-дифторэтана через перегруппировку карбкатионов I→II представляется логичным.
Следует отметить, что такая перегруппировка I→II характерна для всех изучаемых HVMFs, причем 1,2-дифторэтан в продуктах реакций с трифторидом марганца и тетрафторидом церия присутствовал в незначительных количествах. Возможно, это связано с тем, что процессы фторирования трифторидом марганца и тетрафторидом церия проходили при значительно более высоких температурах.
Карбкатион I может появляться в процессе фторирования не только этилена, но и фторэтана (CH2FCH3). В молекуле CH2FCH3 электронная плотность у атома углерода, связанного со фтором, должна быть смещена к атому фтора, следовательно преимущественный отрыв электрона с последующим замещением водорода должен происходить у углерода, не связанного со фтором (Схема 3).

Схема 3.
В работе японских исследователей (S. Kurosawa с сотр.) [13] приведены результаты фторирования фторэтана трифторидом кобальта в периодическом реакторе при температурах 50-100 ºC и времени контакта 8 часов, где наблюдалось соотношение 1,1-дифторэтана к 1,2-дифторэтану ≈2÷4:1, что также может служить определенным подтверждением возможности перегруппировки I→II (Схема 2).
2.4.2. Влияние геминальных атомов фтора на замещение водорода
Полученные результаты подтверждали, что легкость замещения водорода снижалась при наличии геминальных атомов фтора (См. [2]). В качестве примера были приведены результаты фторирования 1,1-дифторэтана трифторидом кобальта и тетрафторидом церия (Табл. 1), которые показывали преимущественное образование 1,1,2-трифторэтана (соотношение 1,1,2-трифторэтана к 1,1,1-трифторэтану составляло ≈10÷20:1 для CoF3 в зависимости от температуры взаимодействия). Для CeF4 соотношение изомеров трифторэтана было еще более значительным (содержание CHF2-CH2F к CF3-CH3 ≈100:1).
Табл. 1. Результаты фторирования 1,1-дифторэтана трифторидом кобальта и тетрафторидом церия (время контакта ~7 минут) [4].
|
T, ºC |
HVMF |
Состав (% об.) |
|||||
|
CHF2-CH3 |
CHF2-CH2F |
CF3-CH3 |
CHF2-CHF2 |
CF3-CH2F |
CF3CHF2 |
||
|
97 |
CoF3 |
14,4 |
73,3 |
7,3 |
1,5 |
3,5 |
0,0 |
|
118 |
CoF3 |
0,1 |
76,0 |
3,1 |
7,5 |
12,1 |
1,2 |
|
290 |
CeF4 |
2,0 |
68,6 |
0,6 |
27,6 |
- |
0,2 |
Полученные результаты были объяснены тем, что взаимодействие молекулы 1,1-дифторэтана с трифторидом кобальта (или другого HVMFs) начиналось с отрыва электрона ионом Co+3 от органического субстрата. В 1,1-дифторэтане электронная плотность у атома углерода, связанного со фтором сильно смещена к атомам фтора, следовательно должен был происходить преимущественный отрыв электрона с последующим отщеплением H+ у углерода, не связанного со фтором (Схема 4) [6].

Схема 4.
На втором этапе радикал IV либо мог подвергаться окислению ионом металла (Co3+) с образованием карбкатиона IV’, либо происходило «гашение» радикала через переход атома фтора из CoF3 [6]. Аналогичные закономерности должны были проявляться и на последующих стадиях фторирования.
2.4.3. Влияние условий проведения процесса и природы HVMFs на соотношение изомеров полифторэтанов
Тезис о том, что замещение водорода на фтор на промежуточных стадиях фторирования происходит преимущественно у атома углерода, который связан с наименьшим количеством атомов фтора хорошо подтверждался результатами фторирования на слабых фторирующих агентах: MnF3 и CeF4 (см. Рис. 5 и 6). Приведенные в работе [12] результаты фторирования на KCoF4 также укладываются в эту концепцию. В то же время при фторировании на CoF3 наблюдалось высокое содержание полифторэтанов с ассиметричным распределением фтора.
Из литературных источников известно, что карбкатионы (или аналогичные радикалы) с положительным зарядом у атома углерода с максимальным количеством атомов фтора являются более энергетически предпочтительными чем их изомеры с положительным зарядом у атома углерода с минимальным количеством атомов фтора. В качестве примера можно привести уже рассмотренную выше (п. 2.1.1) перегруппировку +CH2CH2F→CH3CHF+ (Схема 2), а также результаты работ S. Kurosawa с коллегами [13-16]. В статье [14] приведены расчеты (метод Хартри-Фока, unrestricted Hartree-Fock (UHF) calculation performed on PM3 and ab initio methods) электронной структуры радикалов, которые показали, что радикал CH3CF2• (V, Схема 4) является более энергетически предпочтительным, чем радикал CH2FCH2• (IV, Схема 4). Аналогичные расчеты показали энергетическую предпочтительность [CFH2-CF2•] перед [CF2H-CFH•][15].
В подтверждение расчетных данных в статье [13] были приведены результаты фторирования этана, фторэтана, 1,1-дифторэтана и других полифторэтанов трифторидом кобальта в диапазоне температур 50÷200 °C. Фторирование проводилось в периодическом режиме, время взаимодействия составляло 8 часов (в одном синтезе 400 часов). Результаты фторирования 1,1-дифторэтана трифторидом кобальта из работы приведены в Табл. 2.
Табл. 2. Результаты фторирования 1,1-дифторэтана трифторидом кобальта при времени контакта 8 часов (* время контакта 400 часов) [13].
|
Реактант |
T, °C |
Состав (%) |
|||||
|
CH3CF2H |
CFH2-CFH2 |
CF3CH3 |
CF2H-CFH2 |
CF3CFH2 |
CF2HCFH2 |
||
|
CH3CF2H |
50 |
86 |
0 |
11 |
2 |
1 |
0 |
|
CH3CF2H |
50 * |
4 |
0 |
58 |
12 |
25 |
1 |
|
CH3CF2H |
75 |
53 |
0 |
29 |
14 |
4 |
0 |
|
CH3CF2H |
100 |
6 |
0 |
34 |
49 |
7 |
4 |
При сравнении результатов Табл. 1 и 2 можно отметить, что даже при более низких температурах, но значительно более высоком значении времени взаимодействия наблюдается смещение процесса в сторону образования 1,1,1-трифторэтана, причем увеличение времени реакции до 400 часов приводило к получению 1,1,1-трифторэтана с почти 60% выходом и соотношению изомеров CF3CH3 :CF2H-CFH2≈5:1. Вероятный механизм образования 1,1,1-трифторэтана через перегруппировку с миграцией H+ (или радикала H•) представлен на Схеме 4.
На основании анализа результатов фторирования 1,1-дифторэтана из различных источников можно предположить, что именно при фторировании на CoF3 создавались условия, которые способствовали перегруппировкам карбкатионов (или радикалов) с образованием более энергетически предпочтительных соединений. И одним из факторов, которые могли влиять на такие перегруппировки, было время взаимодействия (контакта) органического соединения с высшим фторидом.
При этом под временем контакта следует понимать обобщенный показатель совокупности процессов, в том числе: сорбция молекулы органического соединений на поверхности фторида, транспортировка фтора на поверхность частицы, химическая реакция, десорбция молекулы органического соединения и т.д.
Гипотеза была экспериментально проверена изучением зависимости соотношений изомеров тетрафторэтана при фторировании этилена на CoF3 при 250 ºC и разных временах контакта (1-7,5 с). Результаты приведены в Табл. 3 [4].
Табл. 3. Состав продуктов и соотношение изомеров тетрафторэтана при T=250 ºC и временах контакта 1÷7,5 с [4].
|
Time (s) |
Состав (об. %) |
||||||||
|
CF2HCH3 (1) |
CFH2CFH2 (2) |
CF3CH3 (3) |
CF2HCFH2 (4) |
CF2HCF2H (5) |
CF3CFH2 (6) |
C2F5H (7) |
C2F6 (8) |
5/6 ratio |
|
|
bp -24,05 |
bp 26 |
bp -47,6 |
bp 5 |
bp -22,5 |
bp -26,5 |
bp -48,5 |
bp -78 |
||
|
1 |
0,5 |
1,0 |
0 |
85,0 |
9,0 |
4,0 |
0,5 |
0 |
2,25:1 |
|
2,5 |
0 |
0,5 |
1,0 |
56,0 |
24,0 |
15,0 |
3,0 |
0,5 |
1,6:1 |
|
7,5 |
0 |
0 |
1,0 |
47,0 |
26,3 |
19,5 |
6,5 |
0,7 |
1,34:1 |
Bp (Boiling Point)- Температура кипения
Эти результаты достаточно хорошо коррелируют с результатами исследований из статьи [7] (Табл. 4)
Табл. 4. Состав продуктов и соотношение изомеров тетрафторэтана при различных температурах и малых временах контакта [7].
|
Т, ºC |
Time (s) |
Состав (об. %) |
||||||||
|
CFH2CFH2 (2) |
CF2HCH3 (1) |
CF3CH3 (3) |
CF2HCFH2 (4) |
CF2HCF2H (5) |
CF3CFH2 (6) |
C2F5H (7) |
C2F6 (8) |
5/6 ratio |
||
|
150 |
1,4 |
19,9 |
8,1 |
0,7 |
70,3 |
1,8 |
0,0 |
0,0 |
0,0 |
|
|
2,8 |
1,0 |
0,3 |
0,9 |
86,9 |
5,6 |
5,0 |
0,3 |
0,0 |
1,12:1 |
|
|
350 |
1,0 |
0,0 |
0,0 |
0,2 |
5,2 |
20,0 |
10,7 |
43,2 |
20,7 |
1,86:1 |
|
2,0 |
0,0 |
0,0 |
0,0 |
1,4 |
10,6 |
6,0 |
40,0 |
42,0 |
1,76:1 |
|
Другие возможные пути появления 1,1,1,2-тетрафторэтана, а именно: реакция отщепления HF от 1,1,2-трифторэтана и изомеризация 1,1,2,2-тетрафторэтана не принимались во внимание по следующим причинам:
Во- первых, отщепление фтористого водорода обычно стимулируется повышением температуры реакции и в изучаемых реакциях должно было бы наблюдаться максимальное содержание 1,1,1,2-тетрафторэтана в реакциях с CeF4, что не соответствовало экспериментальным данным. Кроме того, 1,1,2-трифторэтан является достаточно стабильным соединением вплоть до 300°C, следовательно, в условиях процесса отщепление фтористого водорода от 1,1,2-трифторэтана маловероятно [6].
Во-вторых, экспериментально было установлено, что в процессе фторирования чистого 1,1,2,2-тетрафторэтана трифторидом кобальта в продуктах реакции присутствовали только сам 1,1,2,2-тетрафторэтан, пентафторэтан и гексафторэтан. Т.е. изомеризация не происходила [6].
Полученные результаты показали (Табл. 3 и 4), что с увеличением времени контакта соотношение между изомерами сдвигалось в сторону 1,1,1,2-тетрафторэтана, что может служить определенным подтверждением гипотезы. Увеличение времени контакта до примерно 300-360 с (6-7 мин.) сдвигало соотношение CF2HCF2H к CF3CFH2 до ≈0,6:1.
Механизм, который объяснял появление 1,1,1,2-тетрафторэтана, был предложен в статье [6] и заключался в возможности перегруппировок промежуточных радикала VI (или карбкатиона VII), которые приводили к появлению ассиметричного изомера (Схема 5). Этой же перегруппировкой радикала VI (или карбкатиона VII) можно объяснить появление 1,1,1,2-тетрафторэтана и в продуктах взаимодействия ацетилена с трифторидом кобальта (см. п. 2.2).

Схема 5. Механизм образования 1,1,1,2-тетрафторэтана [6].
Другим параметром, влияющим на соотношение изомеров, была природа фторирующего реагента. Было установлено, что при проведении фторирования на одном и том же оборудовании в сопоставимых условиях (например, при максимальном содержании тетрафторэтанов в продуктах) соотношение концентраций ассиметричного изомера (CF3CFH2) к симметричному (CF2HCF2H) составляло 1,5-1,7:1 для сильного трифторида кобальта, 0,25-0,4:1 для более слабого трифторида марганца, и 0,01-0,02:1 для еще более слабого тетрафторида церия [3,4]. Эти данные хорошо соотносятся с зависимостью силы фторирующего реагента от окислительного потенциала иона металла (см. первую часть сообщения [1]).
CoF3> MnF3> CeF4
Резюмируя вышесказанное, можно с высокой долей вероятности предположить, что применение сильного HVMFs и увеличение времени взаимодействия будут способствовать перегруппировкам промежуточных радикалов (карбкатионов) с образованием ассиметричных полифторалканов. В работах Дж. Бурдона этот процесс также назван H-миграцией (см. [2]). И наоборот, снижение времени контакта в случае сильного HVMFs и применение слабых HVMFs приведет к преимущественному образованию полифторалканов с более равномерным распределением атомов фтора.
Дополнительным фактором, влияющим на распределение изомеров, может являться различная способность к адсорбции симметричных и ассиметричных изомеров-прекурсоров на поверхности трифторида кобальта из-за значительной разницы в температурах кипения (значения Ткип. см. Табл. 3). В работе [7] было высказано предположение, что дифторэтаны и трифторэтаны с более равномерным распределением атомов фтора более склонны к такой адсорбции. Поэтому (по мнению авторов) скорость превращения низкокипящего и плохо адсорбирующегося 1,1-дифторэтана (Ткип≈-24°C) будет определяться скоростью десорбции высококипящих продуктов с поверхности трифторида кобальта. И, соответственно, для 1,2-дифторэтана с относительно высокой Ткип (≈+26°C) и более высокой способностью к адсорбции скорость превращения будет определяться скоростью химической реакции с трифторидом кобальта. Соответственно, скорость дальнейшего превращения 1,2-дифторэтана будет всегда выше скорости превращения 1,1-дифторэтана, поэтому при увеличении времени контакта соотношение изомеров дифторэтана будет смещаться в сторону преобладания 1,1-дифторэтана за счет его более медленного превращения в трифторэтаны. В качестве комментария к этому предположению можно отметить, что способность к адсорбции может влиять на состав продуктов взаимодействия, но только в ограниченном интервале температур и на определенных стадиях процесса, поскольку:
- с увеличением температуры фторирования влияние факторов способности к адсорбции должно снижаться.
- Способность к адсорбции никак не может влиять на соотношение изомеров тетрафторэтана, поскольку фторирование при небольших временах контакта проходит почти исключительно через относительно высококипящий 1,1,2-трифторэтан (Ткип=+5°C).
2.4.4. Механизм образования олигомеров при фторировании тетрафторидом церия
Характерной особенностью взаимодействия этилена с тетрафторидом церия являлось наличие в продуктах взаимодействия в диапазонах температур 150-270°C значительных количеств жидких при нормальных условиях соединений (до 30% по массе от собранных продуктов). Их анализ показал присутствие полифторалканов и полифторалкенов с числом атомов углерода 4 и более.
Была выдвинута гипотеза, что при температурах 150-270°C образование олигомерных соединений протекает по механизму ионной полимеризации, когда образующийся в процессе закрытия двойной связи карбкатион I мог иметь достаточное время жизни, чтобы вступать в конкурирующую реакцию с этиленом с образованием олигомеров (Схема 6) [6]. При более высоких температурах реакция «гашения» карбкатиона фтором (с возможными перегруппировками аналогично Схеме 2) становилась основной.

Схема 6. Механизм образования олигомеров при фторировании этилена тетрафторидом церия [6].
Этот механизм можно объяснить с точки зрения положений катион-радикальной тории фторирования, рассмотренной в первой части сообщения [1]. Направление процессов фторирования определяется временем доставки фтора к поверхности кристаллов HVMFs. Например, в случае фторирования аренов дополнительные пространственные затруднения доставки фтора к поверхности при применении CsTlF4 (и CsCoF4) создавало предпосылки для 1,2-миграции фтора с образованием фторсодержащих ароматических соединений. Фторирование бензола на CeF4 при 480°C также приводило к получению значительного количества фторсодержащих бензолов с 3-4 атомами фтора [17], что также может свидетельствовать о наличии пространственных затруднений доставки фтора к поверхности в кристаллах CeF4. В этом случае появлялась возможность протекания побочной реакции полимеризации с получением олигомерных продуктов. Аналогичный механизм появления олигомеров может быть применен и к фторированию ацетилена на CeF4 (см. п. 2.2).
2.4.5. Возможный механизм образования 1,1,2-трифторэтана на CoF3 при низких температурах
При изучении взаимодействия этилена с трифторидом кобальта при низких температурах (20-60ºC) было обращено внимание на присутствие в продуктах реакций больших количеств (20-30%) 1,1,2-трифторэтана [5]. При этом экспериментально было установлено, что ни 1,2-дифторэтан, ни 1,1-дифторэтана в этом диапазоне температур не взаимодействовали с трифторидом кобальта.
Для объяснения результатов была выдвинута гипотеза, что при низких температурах фторирование на CoF3 могло проходить частично по аналогии с фторированием ароматических соединений. Например, при фторировании бензола трифторидом кобальта процесс проходил через образование фторбензола, 1,4-дифторбензола и только потом начинался процесс присоединения к ароматическому кольцу (подробно рассмотрено в 1-й части [1]). Если предположить, что и в случае фторирования этилена при низких температурах на начальной стадии процесс мог частично проходить через замещение водорода с образованием монофторэтилена с последующим присоединением фтора по двойной связи (Схема 7), то появление больших количеств (20-30%) 1,1,2-трифторэтана становится возможным.

Схема 7.
Выдвинутая гипотеза подтверждается тем, что монофторэтилен был обнаружен в следовых количествах в продуктах реакции этилена с другим высшим фторидом - тетрафторидом церия. В качестве гипотезы возможность реакции замещения была также выдвинута в сообщении [4] при рассмотрении фторирования ацетилена на CeF4 с возможным образованием на первой стадии фторирования монофторацетилена. Однако, следует отметить, что четкие экспериментальные доказательства образования CHF=CH2 при использовании трифторида кобальта отсутствуют.
2.4.6. Возможное влияние фтористого водорода и перехода CoF3→CoF2 на процессы фторирования
Впервые предположение о возможном каталитическом влиянии фтористого водорода на процессы фторирования трифторидом кобальта было сделано в работе [3]. Более подробно влияние HF на фторирование дифторметана, трифторметана и пентафторэтана было изучено в статье [8].
На основании экспериментальных результатов авторы [8] предположили, что в температурном интервале 185-205ºC механизм реакции изменялся и вплоть до 300 ºC фторирование проходило через два конкурирующих механизма. Причиной таких изменений мог служить переход через критическую температуру HF (188ºC) и, как следствие, резкий переход фтороводорода в другое агрегатное состояние даже при незначительном изменении температуры.
Предположения были обоснованы тем, что HF обладает высокой адсорбционной способностью (фториды кобальта при нормальных условиях способны удерживать на поверхности до 0.3 моля HF на 1 моль фторида). Поэтому в процессе фторирования могла появляться тонкая пленка HF на поверхности трифторида кобальта. В этом случае резкое изменение свойств HF при переходе через критическую температуру приводило к десорбции большей части HF с поверхности. После этого поверхность фторидов очищалась и газовые реагенты взаимодействовали непосредственно с трифторидом кобальта. Наблюдаемое уменьшение скорости реакции при переходе через критическую температуру HF авторы объяснили тем, что так как HF является катализатором, то при понижении его концентрации на реакционной поверхности уменьшалась и скорость реакции. Выше 300 ºC фтористый водород полностью десорбировался, и его влияние его на процесс прекращалось.
Был предложен механизм катализа, который заключался в том, адсорбированный фтороводород образовывал водородные связи с адсорбированными молекулами газовых реагентов и поэтому уменьшал акцепторное действие атомов фтора (Схема 8). Это в свою очередь облегчало отрыв электрона от атома углерода с последующим образованием катион-радикала.

Схема 8.
В заключении был сделан вывод, что фтороводород мог участвовать при фторировании одновременно в двух процессах. С одной стороны, адсорбированный на поверхности трифторида кобальта HF мог являться катализатором, с другой - накопление HF на поверхности приводило к диффузионным затруднениям при транспортировке газовых реагентов к реакционной поверхности и к блокированию реакционных центров.
Также в статье [8] на основании экспериментальных данных было показано, что торможение процесса фторирования за счет диффузии газовых реагентов через слой образующегося CoF2 отсутствует. Отсутствие диффузионного торможения было объяснено тем, что поскольку плотность дифторида кобальта выше плотности трифторида, то в твердом слое постоянно образуются трещины, и условия для диффузионных ограничений не создаются.
2.5. Фторирование органических соединений с использованием комплексных фторидов никеля и меди
Фторирование комплексными фторидами никеля (+4) и меди (+3) в реакциях с углеводородами описано в сообщении [9]. Следует отметить, что такие комплексные фториды в отличии от большинства HVMFs могут быть получены из широко распространенных исходных реагентов (хлоридов никеля и меди). K2NiF6 и K3CuF6 были получены непосредственно в реакторе из смесей фторида калия с CuCl или NiCl2 соответственно обработкой элементным фтором.
Фторирующая способность K2NiF6 и K3CuF6 была изучена в процессах с 1,1,1,2- тетрафторэтаном, гексафторпропиленом и некоторыми углеводородами. Авторами был сделан вывод о возможности применения комплексных фторидов для фторирования алканов и алкенов.
Литература
- Kornilov V. V., Fluorination with high valency metal fluorides, cation-radical theory of fluorination of aromatic compounds, Fluorine notes, Iss. 5(162), http://en.notes.fluorine1.ru/public/2025/5_2025/article_1.html, http://dx.doi.org/10.17677/fn20714807.2025.05.01.
- Kornilov V. V., Possible mechanisms of fluorination of aliphatic hydrocarbons with high valency metal fluorides, Fluorine notes, Iss. 6(163), http://en.notes.fluorine1.ru/public/2025/6_2025/article_1.html, http://dx.doi.org/10.17677/fn20714807.2025.06.0.1.
- Asovich V.S., Kornilov V.V., Kostajev R.A., Maximov B.N., Fluorination of methane and its fluoroderivarives by cobalt trifluoride, Zhurnal Prikladnoi Khimii (Russian Journal of Applied Chemistry), Vol. 67, Iss. 1, 1994, p. 103-107.
- Asovich V.S., Kornilov V.V., Kostajev R.A., Mel’nichenko B.A., Maruev A.V., Maximov B.N., Report “Fluorination of hydrocarbons by highest fluorides of cobalt, manganese and cerium”, The International Conference “Chemistry, Technology and Application of Fluorocompounds” (CTAF’94), Saint Petersburg, Russia, May 30- June 03, 1994.
- Asovich V.S., Kornilov V.V., Kostajev R.A., Mel’nichenko B.A., Maruev A.V., Maximov B.N., Fluorination of hydrocarbons by highest fluorides of cobalt, manganese and cerium, Zhurnal Organichnoi Khimii (Russian Journal of Organic Chemistry), Vol. 30, Is. 8, 1994, p. 1221.
- Asovich V.S., Kornilov V.V., Maximov B.N., Fluorination of ethylene by highest fluorides of cobalt, manganese and cerium, Zhurnal Prikladnoi Khimii (Russian Journal of Applied Chemistry), Vol. 67, Iss. 1, 1994, p. 107-111.
- Kostajev R.A., Pashkevich D.A., Desorption of reaction products during the process of Ethylene Fluorination with Cobalt Trifluoride, Zhurnal Prikladnoi Khimii (Russian Journal of Applied Chemistry), Vol. 67, Iss. 10, 1994, pp. 1624-1628; Костяев Р.А, Пашкевич Д.С., Десорбция продуктов реакций в процессе фторирования этилена трифторидом кобальта, ЖПХ, 1994, Т. 67, Вып. 10, с. 1624-1628.
- Kostajev R.A., Pashkevich D.A., The role of hydrogen fluoride and cobalt difluoride in the processes of hydrocarbon fluorination with cobalt trifluoride, Zhurnal Prikladnoi Khimii (Russian Journal of Applied Chemistry), Vol. 67, Iss. 12, 1994, pp. 2012-2016; Костяев Р.А, Пашкевич Д.С., Роль фтороводорода и дифторида кобальта в процессах фторирования углеводородов трифторидом кобальта, ЖПХ, 1994, Т. 67, Вып. 12, с. 2012-2016.
- Maximov B.N., Kornilov V.V., Kostajev R.A., Kosareva L.N., Complex Fluorides of Nickel- and Copper- as a Reagents for Fluorination of Hydrocarbons, The 2nd International Conference “Chemistry, Technology and Application of Fluorocompounds” (CTAF’97), Saint Petersburg, Russia, September 23-26, 1997.
- Kornilov V.V., Kostajev R.A., Maximov B.N., Mel’nichenko B.A., Fiodorova T.E, Fluorination organic compounds by Cobalt Trifluoride. Zhurnal Prikladnoi Khimii (Russian Journal of Applied Chemistry), Vol. 68, Iss 9, 1995, p. 1409-1417.
- Burdon J., Garnier L, Powell R. L., Fluorination of propane and propene over cobalt (III) trifluoride and potassium tetrafluorocobaltate (III), J. Chem. Soc., Perkin Trans. 2, 1996, Iss. 4., pp. 625-631, http://dx.doi.org/10.1039/P29960000625.
- Burdon J.,Knights J., Parsons I, Tatlow J. The fluorination of ethane and ethene over potassium tetrafluoride cobaltate (III) and cobalt trifluoride, Tetrahedron, 1976, V.32, p.1041-1043, https://doi.org/10.1016/S0040-4020(01)83232-4.
- Kurosawa S., Sekiya A., Arimura T., Suga A., The monofluorination of hydrofluorocarbons over cobalt trifluoride, Journal of Fluorine Chemistry, Volume 62, Issue 1, 1993, Pages 69-76, ISSN 0022-1139, https://doi.org/10.1016/S0022-1139(00)80082-2.
- Kurosawa S., Sekiya A., Arimura T., Suga A., Ab Initio Molecular Orbital Study on Monofluorination Reaction of Hydrofluorocarbons Over Cobalt Trifluoride, Journal of Japan Oil Chemists' Society, 1994, p. 650-652, DOI:10.5650/jos1956.43.650.
- Kurosawa S., Amimura T. Theoretical study of monofluorination reaction, Chem. Express., 1992, Vol.7, Iss. 6, p.429.
- Kurosawa S., Synthesis and Application of Fluorine-Containing Compounds, Journal of Japan Oil Chemists' Society, 1999, Vol. 48, Iss. 11, p. 1247, DOI: 10.5650/jos1996.48.1247
- Hudson A.G., Pedler A.E., Tatlow J.C., The fluorination of benzene over cerium tetrafluoride, Tetrahedron, 1969, Vol. 25, Iss. 18, pp. 4371-4374, https://doi.org/10.1016/S0040-4020(01)82976-8.
Статья рекомендована к публикации к.х.н. М.А. Манаенковой
Fluorine Notes, 2026, 164, 1-2


