Поступило в редакцию: Март 2025
УДК 547.22, 661.48
Fluorine Notes, 2025, 159, 1-2
МЕТОДЫ ПОЛУЧЕНИЯ ФТОРСОДЕРЖАЩИХ КАРБОНОВЫХ КИСЛОТ.
СООБЩЕНИЕ 1
С.В. Вершилова, В.В. Корнилов, А.С.Цырульниковаа, Н.В.Лебедева
а ФГБУ НИИСК
Аннотация: В настоящей части обзора рассмотрены следующие методы синтеза фторсодержащих карбоновых кислот: электрохимическое фторирование (ЭХФ), изомеризация и анионная полимеризация окисей перфторалкенов, олигомеризация перфторалкенов в процессах жидкофазного окисления.
Ключевые слова: электрохимическое фторирование, окиси перфторалкенов, перфторалкены, жидкофазное окисление, анионная полимеризация.
Введение
В настоящем обзоре рассмотрены некоторые методы синтеза фторсодержащих карбоновых кислот. Несмотря на то, что в последние годы произошел отказ от применения перфторированных алифатических карбоновых кислот в качестве эмульгаторов в синтезах фторированных полимеров и сополимеров методом водоэмульсионной полимеризации, фторсодержащие карбоновые кислоты и их производные остаются важными прекурсорами для синтеза разнообразных фторсодержащих материалов.
Помимо традиционных областей применения фторсодержащих карбоновых кислот и их производных (пенообразователи, защитные покрытия, присадки к смазочным материалам и др.) соединения данного класса используют в качестве добавок к электролитам в литий-ионных батареях для улучшения характеристик источников тока данного типа при низких температурах [1]. Фторсодержащие карбоновые кислоты также находят применение в синтезах биологически активных соединений с фторсодержащими фрагментами, которые позволяют улучшить физико-химические свойства (кислотность, липофильность, проницаемость) фармацевтических субстанций [2].
Электрохимическое фторирование (ЭХФ)
Методом ЭХФ получают фторангидриды перфторированных карбоновых кислот, которые гидролизом превращают в соответствующие кислоты. В течение долгого времени метод ЭХФ являлся одним из основных промышленных методов получения этих соединений.
В значительной степени это было связано с производством перфторгептановой (перфторэнантовой), перфтороктановой (PFOA) и перфторнонановой (перфторпеларгоновой) кислот, которые применялись в процессах водоэмульсионной полимеризации для получения политетрафторэтилена (PTFE) и других фторсодержащих полимеров, и сополимеров. В последнее десятилетие из-за требований экологии произошел отказ многих производителей фторполимеров от применения PFOA и ее гомологов с числом атомов углерода более 6 в процессах полимеризации, что несколько снизило значение метода ЭХФ.
Способ получения перфторкарбоновых кислот через ЭХФ достаточно подробно описан в литературе, например в обзорных статьях [3-5].
Механизм процесса ЭХФ подробно изложен в работе [6], в которой было объяснено распределение продуктов на примерах ЭХФ фторангидридов и хлорангидридов изомеров бутановой кислоты (iso-butyryl chloride и др.).
Для синтеза кислот с числом атомов углерода в цепи не более 4-х в качестве исходного сырья применяют галогенангидриды (фтор, хлор) или сложные эфиры соответствующих углеводородных кислот. С увеличением числа атомов углерода в цепи выход линейных продуктов снижается, а основными продуктами ЭХФ становятся перфторированные циклические эфиры (Схема 1) [3].
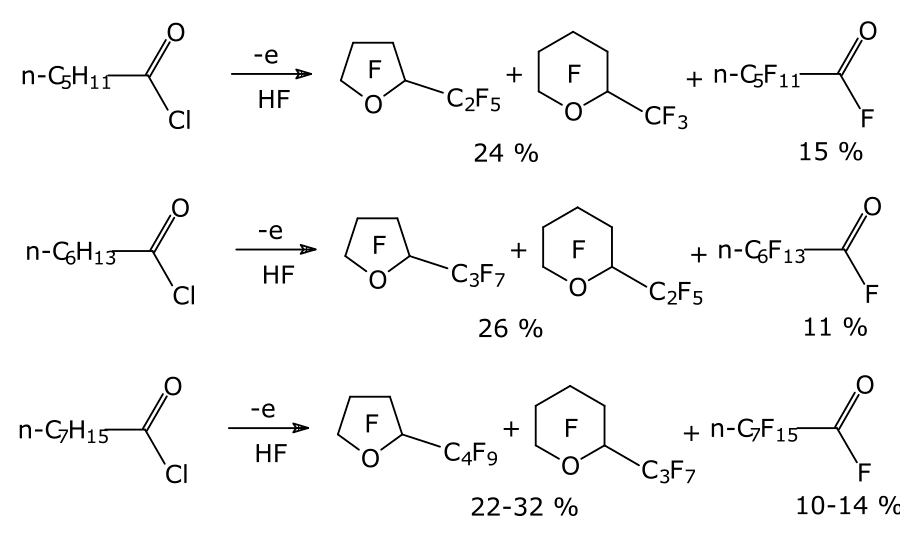
Схема 1.
Поэтому для получения линейных перфторированных карбоновых кислот с более высоким числом атомов углерода в цепи для ЭХФ в качестве исходного сырья использовали соединения уже содержащие атомы фтора.
Один из способов заключается в применении в качестве исходного сырья соответствующих спиртов-теломеров общей формулы H(CF2-CF2)n-CH2-OH (n=2÷5), которые получают в промышленном масштабе взаимодействием тетрафторэтилена с метанолом. Таким методом получали перфторированные пентановую, гептановую, нонановую и ундекановую кислоты с достаточно высоким выходом [7].
Другой способ заключается в предварительном синтезе соединений с линейной углеродной цепью, содержащих в своем составе фторуглеродный фрагмент. В качестве примера можно привести синтез хлорангидрида нонафтороктановой кислоты (4-(perfluoro-n-butyl)-n-butanoyl chloride), который описан в статье G.P. Gambaretto с сотр. (Схема 2) [8].
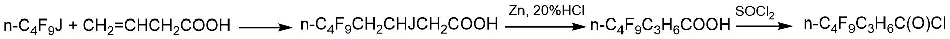
Схема 2.
Последующее ЭХФ позволяло увеличить выход фторангидрида перфтороктановой кислоты до 28% по сравнению с выходом 12,9% при ЭХФ хлорангидрида н-октановой кислоты.
Применение в качестве сырья при ЭХФ фторангидрида нонафтороктановой кислоты (4-(perfluoro-n-butyl)-n-butanoyl fluoride) позволяло увеличить выход фторангидрида перфтороктановой кислоты до 29% [5].
Увеличение выхода целевого линейного продукта при применении частично фторированного сырья было объяснено тем, что в данном случае происходило блокирование процессов образования циклических соединений в ходе ЭХФ.
При получении карбоновых кислот методом ЭХФ требуется поиск условий для каждого такого процесса, включая подбор исходного соединения и его концентрации в электролите.
Например, для получения фторангидрида перфторпропионовой кислоты в качестве исходного сырья традиционно использовались пропионилхлорид или спирт-теломер n=1 (HCF2-CF2-CH2-OH). В работе [9] для синтеза фторангидрида перфторпропионовой кислоты предлагалось использовать более распространенный малеиновый ангидрид для получения с высоким выходом фторангидрида перфторпропионовой кислоты и одновременно дифторангидрида тетрафторянтарной кислоты (Схема 3).
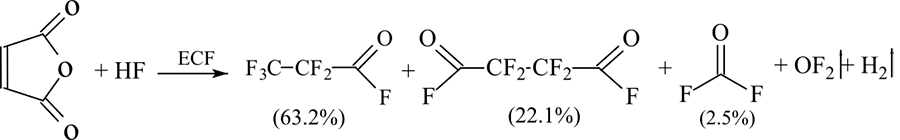
Схема 3.
В ряде случаев используют введение так называемых «активных добавок», которые позволяют стабилизировать режим ЭХФ и вести процесс длительное время. В качестве таких добавок обычно применяют серосодержащие соединения (в частности н-бутилмеркаптан) или третичные амины [10, 11].
2. Изомеризация и анионная полимеризация окисей перфторалкенов, олигомеризация перфторалкенов в процессах жидкофазного окисления
2.1. Изомеризация и анионная полимеризация окисей перфторалкенов
Ряд нуклеофильных фторсодержащих реагентов способен раскрывать эпоксидные кольца в альфа-окисях перфторалкенов с образованием фторангидридных групп. Так, присутствие фторид-ионов способствует быстрому раскрытию этих колец с сохранением перфторированной природы продуктов. Нуклеофильная атака происходит исключительно по более замещенному углероду c образованием перфтоалкоксидов, которые можно в ряде случаев выделить из продуктов реакции [12]. В качестве примеров в работе [12] приведены алкоксиды, полученные из окисей гексафторпропилена (HFPO) и окиси октафтор-изо-бутилена (OFIBO).
|
CF3CF2CF2O-M+(HFPO) |
(CF3)2CFCF2O-M+ (OFIBO) |
При определенных условиях эти соли теряют фрагменты M+ и F- с образованием фторангидридов перфторкарбоновых кислот. Последние с помощью гидролиза могут быть превращены в соответствующие кислоты.
Природа фторид-иона и природа иона M+ играют очень важную роль, как в раскрытии эпоксидного кольца, так и в устойчивости образующегося перфторалкоксида. Авторы работы [12] провели исследования более 40 солей фторидов в различных растворителях и установили, что наиболее лучшие результаты показывал фторид цезия в тетраглиме.
Образующиеся алкоксиды могут вступать в дальнейшую реакцию с молекулами окиси перфторалкена с получением олигомерных продуктов (Схема 4) [12].
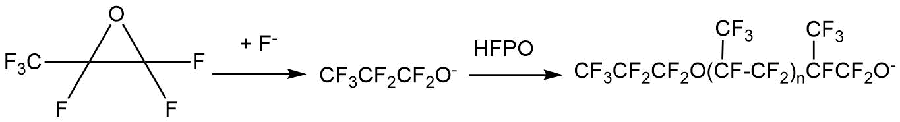
Схема 4.
Кроме того, окиси перфторалкенов могут взаимодействовать с другими первичными фторангидридами состава R-C(O)-F и фторсодержащими кетонами с образованием соответствующих фторангидридов карбоновых кислот.
В литературных источниках описаны реакции окисей тетрафторэтилена (TFEO), гексафторпропилена (HFPO) и некоторых других окисей перфторалкенов.
2.1.1. Изомеризация и анионная полимеризация окиси тетрафтоэтилена (TFEO)
В 1966 г. в статье Ф. Гоцци и Г. Камагги было сообщено об образовании фторангидрида трифторуксусной кислоты из окиси тетрафторэтилена в присутствии фторида калия (TFEO) [13].
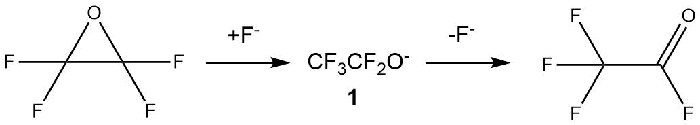
Схема 5.
В 1967 г. В.И. Гинзбург и М.И. Васильева в своей работе также сообщали о перегруппировке TFEO в фторангидрид трифторуксусной кислоты в присутствии фтористого водорода даже при -78ºС [14].
В статье H. S. Eleuterio [15] было сообщено, что перегруппировка TFEO в фторангидрид трифторуксусной кислоты может быть осуществлена при комнатной температуре на поверхности катализатора или в присутствии источников фторид-иона (F-). Кроме того, было отмечено, что реакция могла самопроизвольно происходить даже при комнатной температуре, поэтому TFEO следует хранить только при температурах ниже температуры кипения (-65 ºС).
Как уже отмечалось выше образующийся алкоксид 1 (Схема 5) может вступать в дальнейшую реакцию с молекулами окиси перфторалкена с получением продуктов присоединения.
В патенте [16] приведен пример такой реакции, которую осуществляли с использованием активированного угля “Darco” в качестве катализатора при температурах -52÷-30 ºС. В продуктах реакции были идентифицированы перфторэтоксиацетил фторид (perfluoroethoxyacetyl fluoride, CF2CF2-O-CF2-C(O)F), а также продукты дальнейшего присоединения: тример и тетрамер TFEO.
Полученный тример TFEO мог быть гидролизован водой до соответствующей кислоты CF3-CF2-O-CF2CF2-O-CF2-COOH [16].
Другие примеры получения полимерных соединений TFEO приведены в [17]. Процесс проводили при пониженных температурах (-70÷-80 ºС) в присутствии катализаторов. В качестве катализаторов использовали третичные амины (триэтиламин, трибутиламин и др.), а также соли двухвалентной меди (Cu2+). Состав продуктов зависел от времени синтеза: после 24 часов синтеза в продуктах преобладали жидкие соединения, после 48 часов синтеза – твердые соединения.
В патенте [18] описаны реакции присоединения TFEO в присутствии катализаторов к другим фторангидридам: карбонил фторид, фторангидрид трифторуксусной кислоты, перфторпропионил фторид и др. Реакции проводили в среде метиленхлорида, дихлорметана и различных линейных хлорфторуглеводородов (Схема 6).

Схема 6 (Rf = F, CF3, C2F5 и др., n ≥0).
В качестве катализаторов применяли цианид или бромид тетраэтиламмония.
Так, при взаимодействии фторангидрида трифторуксусной кислоты (99 г) с TFEO (326 г) при температуре -30 ºС и давлении ≈100÷138 кПа, были получены продукты, которые представлены в Табл. 1 (катализатор - цианид тетраэтиламмония) [18].
Табл. 1. Состав продуктов реакции фторангидрида трифторуксусной кислоты с TFEO.
|
Структура |
Вес, г |
Температура кипения, ºС |
|
C2F5-O-CF2COF |
15 |
0-6 |
|
C2F5-O-C2F4-O-CF2COF |
69 |
65-67 |
|
C2F5-O-(C2F4-O)2-CF2COF |
171 |
99-102 |
|
C2F5-O-(C2F4-O)3-CF2COF |
103 |
134-138 |
|
C2F5-O-(C2F4-O)4-CF2COF |
33 |
167-170 |
Аналогично протекали реакции взаимодействия с дифторангидридами фторсодержащих карбоновых кислот. При этом получали бис-фторангидриды, содержащие в цепи простые эфирные связи (Схема 7) [14].
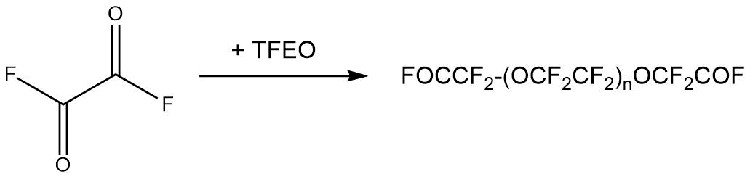
Схема 7 (n =0÷3[17]).
Перфторкетоны также могут взаимодействовать с TFEO с получением соответствующих фтоангидридов карбоновых кислот. В [18] описано взаимодействие гексафторацетона с TFEO с получением фторангидридов общей формулы (CF3)2-CF-O-(C2F4-O)n-CF2COF (n=0÷4) с преимущественным образованием (CF3)2-CF-O-CF2COF.
2.1.2. Изомеризация и анионная полимеризация окиси гексафторпропилена (HFPO)
Реакции окиси гексафторпропилена (HFPO) гораздо более хорошо изучены по сравнению с TFEO, поскольку методы получения HFPO более доступны. Кроме того, HFPO выдерживает высокие температуры до 150 ºС включительно [15].
Хорошо изучен процесс изомеризации окиси гексафторпропилена (HFPO) в реакциях с нуклеофильными агентами с образованием фторангидрида перфторпропионовой кислоты 2 (Схема 8).
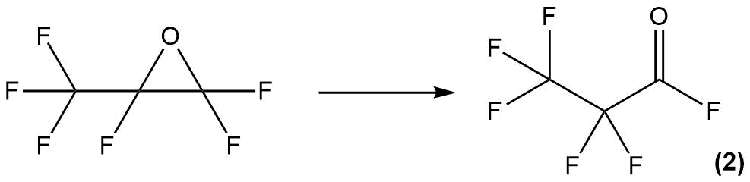
Схема 8.
Для проведения данной реакции могут быть использованы различные нуклеофилы, а именно: первичные, вторичные и третичные амины (в частности, триэтиламин), Me3SiOMe, Me2Si(OEt)2 и др. [19].
Существуют разные версии механизма изомеризации HFPO, которые рассмотрены в работах [19-22].
Взаимодействием HFPO с первичными ацилфторидами состава R-C(O)-F (3) могут быть получены фторангидриды карбоновых кислот общей формулы 5 (Схема 9) [19].
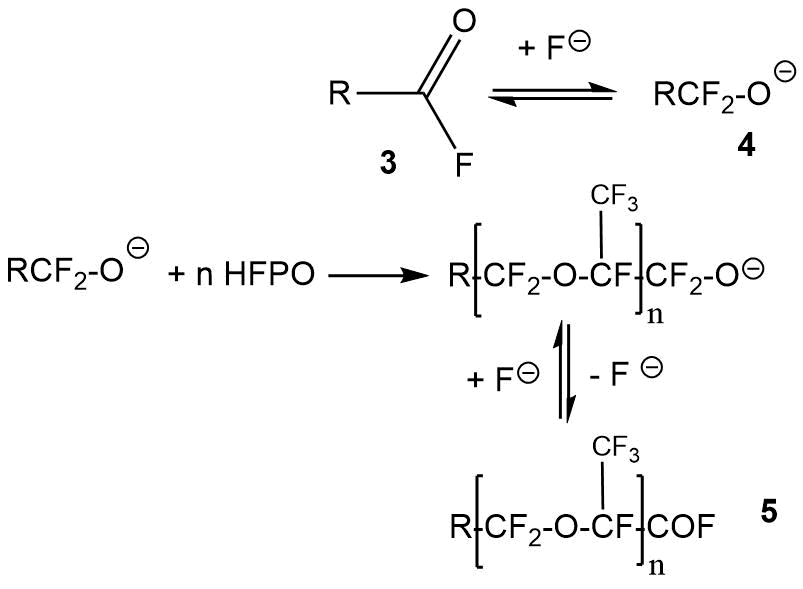
Схема 9.
Исходные фторангидриды 3 могут быть различной структуры. Выход продуктов общей формулы 5 зависит от природы радикала R в фторангидриде 3 и стабильности фторалкоксида 4. Большое количество ссылок на примеры таких реакций приведено в работе [19].
Применение специальных каталитических систем способствует образованию фторалкоксидов 4. Кроме того, применение таких катализаторов должно подавлять образование фторангидрида перфторпропионовой кислоты для предотвращения побочной реакции анионной полимеризации HFPO.
Анионная полимеризация HFPO является частным случаем взаимодействия HFPO с первичными фторалкоксидами и приводит к получению олигомерных фторангидридов 8 с числом n=1÷100 (Схема 10) [19].
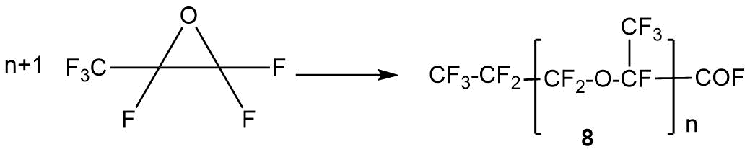
Схема 10.
Первым шагом этого процесса является изомеризация HFPO в фторангидрид перфторпропионовой кислоты (Схема 4), который находится в равновесии с перфторпропоксидным ионом 6 (Схема 11). Последний атакует одну молекулу HFPO с образованием алкоксида 7 с n=1. На следующем этапе алкоксид 7 может далее реагировать с молекулами HFPO с получением алкоксидов с n>1 или образовывать фторангидрид 8 после отщепления фторид-иона [19].
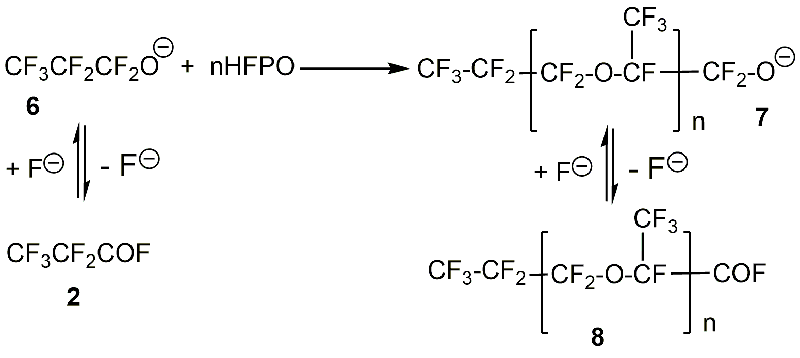
Схема 11.
Степень олигомеризации зависит от используемой каталитической системы, условий проведения реакции и чистоты исходного HFPO [19].
Так, при использовании в качестве катализатора тетраметилтиомочевины ((Me2N)2CS, tetramethyl thiourea) в диглиме были получена смесь олигомеров следующего состава: 13% с n=1, 40% с n=2 и 22% с n=3 [23].
В Табл. 2 приведены примеры зависимости степени олигомеризации HFPO от природы растворителя [19].
Таблица 2. Зависимость степени олигомеризации HFPO (n) от природы растворителя.
|
Растворитель |
Выход (%) |
||||
|
n=0 |
n=1 |
n=2 |
n=3 |
n=4 |
|
|
Тетрагидрофуран |
100 |
||||
|
Ацетонитрил |
88 |
9 |
3 |
||
|
Моноглим |
71 |
20 |
9 |
||
|
Диглим |
32 |
31 |
25 |
10 |
|
|
Тетраглим |
16 |
17 |
21 |
20 |
10 |
Реакции HFPO с фторсодержащими кетонами приводят к получению фторангидридов фторсодержащих карбоновых кислот общей формулы 12 (Схема 12) [19].
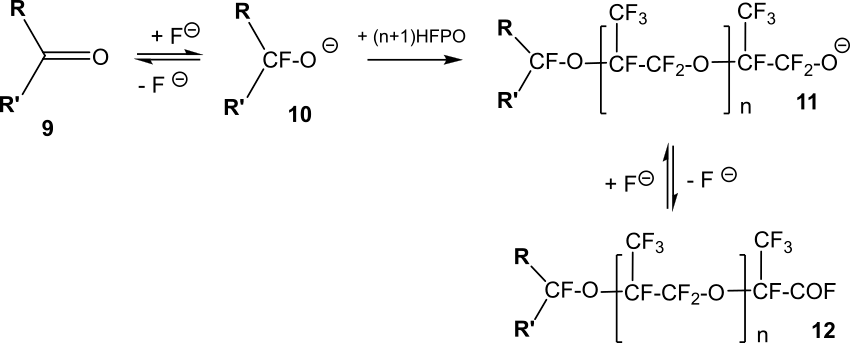
Схема 12.
Вторичные фторированные алкоксиды 10, которые получаются в присутствии фторид-ионов из фторсодержащих кетонов 9, в результате взаимодействия с HFPO образуют алкоксиды общей формулы 11 (n=0). Дальнейший процесс идет либо с отщеплением фторид иона с образованием фторангидрида 12 (n=0), либо с дальнейшим присоединением молекул HFPO к алкоксиду 11 с получением фторангидридов 12 (n≥1).
В патенте [24] описано взаимодействие HFPO с гексафторацетоном (HFA), 1,3-дигидроперфторацетоном, перфтор-2-пентаноном и рядом других перфторкетонов. Условия процессов были следующие: начальная температура -80 ºС с последующим нагревом до 80 ºС, катализатор – фторид цезия в диглиме. В результате реакций были получены соответствующие фторангидриды общей формулы 12.
В случае реакции HFPO с HFA при соотношении реагентов 1:1 преимущественно был получен перфтор-2-изопропоксипропионил фторид из которого на следующей стадии посредством гидролиза была получена соответствующая кислота. Если же соотношение HFPO к HFA составляло 5:1, то в продуктах реакции 13 присутствовали продукты с n=1-6.
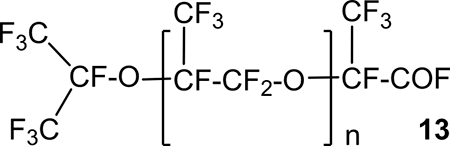
Примеры других синтезов с различными радикалами R и R’ в кетонах 9 (Схема 12), а также условия взаимодействия соединений 9 с HFPO (соотношение реагентов, катализаторы) приведены также в работе [19].
2.2. Олигомеризация перфторалкенов в процессах жидкофазного окисления
Жидкофазное окисление перфторолефинов используют в промышленности для получения перфторполиэфиров (ПФПЭ), которые известны c 1960-х гг. под торговой маркой Fomblin® (Solvay).
Процесс окисления тетрафторэтилена (TFE) и гесафторпропилена (HFP) проводят молекулярным кислородом при пониженной температуре в присутствии инициатора с получением олигомерных продуктов приблизительной структуры 14 и 15 (пероксидные ПФПЭ, ПО ПФПЭ) на Схеме 13 соответственно.
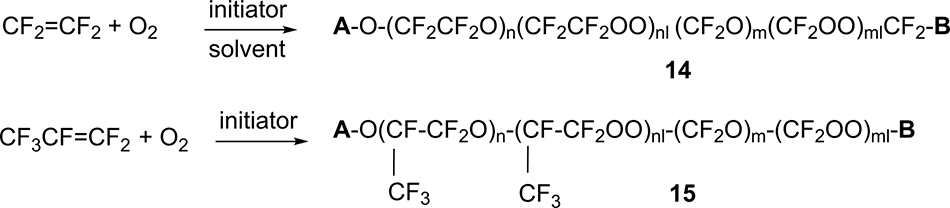
Схема 13.
В случае жидкофазного окисления HFP в качестве жидкой фазы выступает сам HFP, процессы окисления TFE требуют применения инертного растворителя.
В качестве инициаторов используют либо УФ-облучение, либо химические реагенты: элементный фтор, трифторметилгипофторит и др.
Группы А и В на схеме 13 являются перфторалкильными или ацилфторидными (в большинстве случаев группы -CF3, -CF2CF3, -COF или -CF2COF).
Например, в случае жидкофазного окисления HFP при использовании в качестве инициатора УФ-облучения соотношение групп A и B было следующим: перфторалкил-ацилфторид: перфторалкил-перфторалкил: ацилфторид- ацилфторид = 2:1:1 [25].
В получаемых ПО ПФПЭ 14 и 15 присутствуют фрагменты со связями (-O-O-), удаление которых происходит на следующей стадии синтеза.
Сами процессы получения ПФПЭ достаточно полно описаны в литературе. Основные закономерности жидкофазного окисления HFP и TFE при инициировании УФ-облучением обобщены в работе Д. Сианези с сотр. [25]. Процессы жидкофазного окисления HFP с химическим инициированием (элементный фтор) описаны, например, в [26, 27]. Особенности процессов жидкофазного окисления TFE с использованием различных химических инициаторов, а также механизм и кинетические закономерности процессов приведены в статьях [28-32]. Кроме того, существует большое количество патентной литературы.
Для удаления пероксидных фрагментов (-O-O-) применяют термическую обработку в диапазоне температур 150÷300ºC, термическую стабилизацию с одновременным УФ облучением [25] или химические методы [32-34].
В результате получают олигомерные соединения 16 и 17 (Схема 14) в большинстве случаев имеющие в своей структуре хотя бы одну фторангидридную группу, которая может быть переведена гидролизом в соответствующую кислоту.
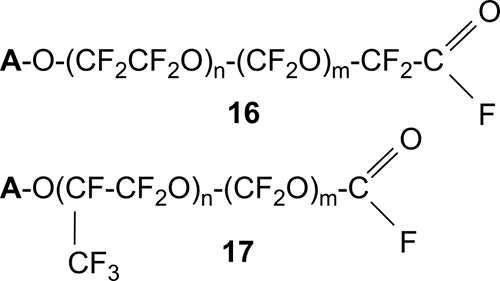
Схема 14.
Изменяя параметры проведения процессов (температура, соотношение реагентов, природа инициатора, метод удаления перокидных групп и т.д.), можно регулировать длину цепи получаемых соединений и/или преимущественный состав оконечных групп.
Например, проведение жидкофазного окисления HFP при температуре около -30ºС (инициатор – F2) приводило к снижению молекулярной массы олигомера 15 до ММ ≈ 1000 [27], тогда как проведение процесса при -50÷-60 ºС приводит к получению олигомера 15 с ММ≈3000÷3500.
В работе [33] описано получение пероксидного ПФПЭ низкотемпературным жидкофазным окислением TFE (УФ-облучение, T=-40ºС, растворитель - R227ea) с одновременной добавкой в реактор перфторакролоил фторида (CF2=CF-COF). В этом случае получали ПО ПФПЭ (ММ≈2900) общей формулы X1-O(CF2O)n1(CF2CF2O)m1(CF2(CF2)zCF2O)p1 (O)h-X2, где группы X1 и X2 были -CF2-COF или -COF. Разрушение пероксидных связей с помощью 57% водного раствора иодистоводородной (HI) кислоты в присутствии метанола привело к образованию продуктов общей формулы Y1-O(CF2O)n1(CF2CF2O)m1(CF2(CF2)zCF2O)p1-Y2 со средней MM≈490, где Y1 и Y2 были группами -CF2COOCH3 и/или -CF2COOH.
В работе [34] также описано разрушение пероксидных связей в ПО ПФПЭ с помощью 57% водного раствора иодистоводородной (HI) кислоты в присутствии метанола. Однако, исходный ПО ПФПЭ был получен низкотемпературным жидкофазным окислением TFE (инициатор – F2, T=-82 ºС, растворитель дихлордифторметан) без применения дополнительных ингредиентов. В этом случае получали продукты общей формулы Y-O(CF2O)a(CF2CF2O)b-Y’ со средней MM≈930, где Y и Y’ были группами -CF2COOCH3, -CF3, -C2F5 с существенным преобладанием сложно-эфирной группы.
В статье [32] описано низкотемпературное жидкофазное окисление HFP, когда в реактор кроме мономера, кислорода и инициатора вводили в качестве «сомономера» хлортрифторэтилен (CTFE). В результате были получены полиэфирные теломеры общей формулы Cl–(C3F6O)n–CF2CFO. Полученную смесь можно было разделить на индивидуальные соединения дистилляцией. Полученные полиэфирные теломеры с выходом более 85% (вес.) были переведены в соответствующие карбоновые кислоты: Cl–(C3F6O)n–CF2COOH, где n=1÷5.
Процесс получения полиэфирных бис-фторангидридов общей формулы 16 (группа A на схеме 14 также преимущественно была ацилфторидной) описан в [35]. Исходный пероксидный ПФПЭ 14 (Схема 14), который был получен низкотемпературным жидкофазным окислением ТФЭ со средней ММ≈83400 и имел в своем составе преимущественно одну фторангидридную группу, был обработан газообразным водородом в присутствии катализатора (Pd/CaF2) при температуре 125÷-130 ºС и давлении около 3 атм. Полученные бис-фторангидриды не содержали пероксидных связей и имели ММ≈1500.
В работах [26,36] сообщается о получении из фторангидридов общей формулы 17 (n=8÷50, m=0÷10) соответствующих кислот, которые могут быть далее использованы для получения разнообразных ПАВ.
Список Литературы
- Jinhui Han, Xiaohua Yang, Zhiping Liu, Effect of Fluorinated Carboxylic Acid Ester on Lithium Solvation as an Additive in Electrolyte and Low-Temperature Insight on Battery Performance, Ind. Eng. Chem. Res. 2023, 62, 19, 7682–7692, https://doi.org/10.1021/acs.iecr.3c00417.
- Thibault Alle, Sipak Joyasawal, Killian Oukoloff, Keith Long, Zachary Owyang, Karol R. Francisco, Dominique Cahard, Donna M. Huryn, Carlo Ballatore, Structure-property relationships of fluorinated carboxylic acid bioisosteres, Bioorg. Med. Chem. Lett. 91 (2023), 129363, https://doi.org/10.1016/j.bmcl.2023.129363.
- G.G.Furin, Perfluorinated Carboxylic Acids. Synthesis and Application, Fluorine Notes, Iss. 3(34) 2004 (http://en.notes.fluorine1.ru/contents/history/2004/3_2004/retro/index.html).
- V. М. Berenblit, Electrochemical fluorination of organic compounds, Fluorine Notes, Iss. 1(2)-3(4), 1999 (http://notes.fluorine1.ru/contents/history/1999/1_1999/Notes/retro/index.html).
- Alsmeyer, Y.W., Childs, W.V., Flynn, R.M., Moore, G.G.I., Smeltzer, J.C. (1994). Electrochemical Fluorination and Its Applications. In: Banks, R.E., Smart, B.E., Tatlow, J.C. (eds) Organofluorine Chemistry. Topics in Applied Chemistry. Springer, Boston, MA. DOI: 10.1007/978-1-4899-1202-2_5.
- N.V. Ignat’ev, Electrochemical Fluorination: A Powerful Tool for the Preparation of Organofluorine Compounds. In book: Modern Synthesis Processes and Reactivity of Fluorinated Compounds, pp.71-123, 2017. DOI: 10.1016/B978-0-12-803740-9.00004-4
- B. N. Maksimov, L.N. Kosareva, N.A. Rjabinin, Method of preparing higher perfluoromonocarboxylic acids, Patent RU 2107751, 1993.
- M. Napoli, L. Conte, G.P. Gambaretto, F.M. Carlini, Electrochemical fluorination of 4-(perfluoro-n-butyl)-n-butanoyl chloride, Journal of Fluorine Chemistry, Volume 45, Issue 2, 1989, Pages 213-224, DOI: 10.1016/S0022-1139(00)84147-0
- N.B. Lesnevskaya, E.V. Litvinenko, A.A. Ludikainen, V.A. Matalin, T.V. Mikhailova, Electrochemical fluorination of maleic anhydride, Fluorine Notes, Iss. 6(121) 2018 (http://en.notes.fluorine1.ru/public/2018/6_2018/article_2.html), DOI 10.17677/fn20714807.2018.06.02.
- Pat. US3028321, Electrochemical production of fluorocarbon acid fluorides, 1962.
- G.I. Kaurova, V.A. Matalin, A.A. Krasil’nikov, V.G. Barabanov, B.N. Maksimov, Electrochemical Fluorination in Synthesis of Fluoroorganic compounds, In book: V.I. Manuylova, B.N. Maximov, V.V. Kornilov (Eds.) "Fluorine compounds. Chemistry, Technology, Application", Teza, Saint-Petersburg, 2009, 356 p., ISBN 5-88851-068-8, pp.69-82 (in Russian).
- J. T. Hill and J. P. Erdman, Anionic Polymerization of Fluorocarbon Epoxides, ACS Symposium Series Vol. 59, 1977, pp. 269-284, DOI: 10.1021/bk-1977-0059.ch019.
- F. Gozzo, G. Camaggi, Oxidation reactions of tetrafluoroethylene and their products-I: Auto-oxidation, Tetrahedron, 1966, Vol. 22, pp. 1765-1770.
- P. Tarrant and E. Stamp, Zh. Vses. Khim. O-va.,1970, 15, 34.
- H. S. Eleuterio, Polymerization of Perfluoro Epoxides. Journal of Macromolecular Science: Part A - Chemistry, 1972, 6(6), pp. 1027–1052, DOI:10.1080/10601327208056884.
- US Patent 3125599, Polymers of fluorocarbon epoxides, 1964.
- US Patent US3380937, Process for polymerizing tetrafluoroethylene epoxide, 1968.
- US Patent 3250806, Fluorocarbon ethers of tetrafluoroethylene epoxide, 1966.
- H. Millauer, W. Schwertfeger, G. Siegemund, Hexafluoropropene Oxide - A Key Compound in Organofluorine Chemistry, Angew. Chem. Inr. Ed. Engl., 1985, Vol. 24, Iss. 3, pp. 161-179, DOI: 10.1002/anie.198501611.
- D. Sianesi, A. Pasetti, F. Tarli, J. Org. Chem., 31, 1966, p. 2312.
- I. L. Knunyants, V. V. Shokina, I. V. Galakhov, Khim. Geterotsikl. Soedin. 1966, p. 873.
- R. A. Bekker, G. V. Asratyan, B. L. Dyatkin, Zh. Org. Khim., 1973, 9, p. 1644.
- US Patent 4118421, Process for the manufacture of perfluoro-alkoxy-propionic acid fluorides, 1978.
- US Patent 3274239, Fluorocarbon ethers, 1966.
- Sianesi, D., Marchionni, G., De Pasquale, R.J. Perfluoropolyethers (PFPEs) from Perfluoroolefin Photooxidation. In: Banks, R.E., Smart, B.E., Tatlow, J.C. (eds) Organofluorine Chemistry. Topics in Applied Chemistry. Springer, Boston, 1994, MA. DOI: 10.1007/978-1-4899-1202-2_21.
- B. N. Maksimov, V. V. Kornilov, B. A. Melnychenko, and L. N. Kosareva, Perfluoropolyethers. Synthesis and Application, Russian Journal of Applied Chemistry, 2009, Vol. 82, No. 9, pp. 1706−1710.
- RU Patent 2046127, Process for preparing polyperfluoropropylene oxide, 1995
- P.A. Guarda, E. Barchiesi, G. Fontana, S. Petricci, M. Pianca, G. Marchionni, Peroxidic perfluoropolyether from tetrafluoroethylene oxidation: micro structural analysis by NMR spectroscopy and mechanistic considerations, Journal of Fluorine Chemistry, 2005, Vol. 126, Iss. 1, pp 141-153, doi:10.1016/j.jfluchem.2004.11.012.
- P.A. Guarda, E. Barchiesi, G. Fontana, S. Petricci, M. Pianca, G. Marchionni, Peroxidic perfluoropolyether from tetrafluoroethylene oxidation: micro structural analysis by NMR spectroscopy and mechanistic considerations, Journal of Fluorine Chemistry, 2005, Vol. 126, Iss. 4, Pp. 683-695, DOI: 10.1016/j.jfluchem.2005.01.002.
- D. Sianesi, P. A. Guarda, G. Marchionni, A Kinetic and Mechanistic Study of the Low-Temperature Fluorine-Initiated Copolymerization of Tetrafluoroethylene with Oxygen, Macromolecules, 1999, Vol. 32, Iss. 23, pp. 7768-7780, DOI: 10.1021/ma9906985.
- P.A. Guarda, S. Petricci, G. Fontana, G. Marchionni, Synthesis and characterization of poly(tetrafluoroethyleneperoxides), Journal of Fluorine Chemistry, 2009, Vol.130, Iss. 11, pp. 1011-1016, DOI: 10.1016/j.jfluchem.2009.07.021.
- Massimo Malavasia, Dario Sianesi, Novelties and prospects in the synthesis of perfluoropolyethers by oxidative polymerization of fluoroolefins, Journal of Fluorine Chemistry, 1999, 95, pp. 19–25.
- EP 1568730, Peroxidic perfluoropolyethers, 2011.
- EP 0654493, Process for preparing perfluoropolyethers, 1995.
- EP 1388555, A process for the preparation of perfluoropolyethers acyl-fluoride ended by reduction of the corresponding peroxidic perfluoropolyethers, 2004.
- RU Patent 2069673, Antifriction composition for treatment of solid surfaces, 1996.
Статья рекомендована к публикации членом редколлегии к.х.н. А.А. Тютюновым
Fluorine Notes, 2025, 159, 1-2


