Поступило в редакцию: Октябрь 2023
УДК 547.32.32, 547
Fluorine Notes, 2023, 150, 5-6
ВЗАИМОДЕЙСТВИЕ ПОЛИФТОРКЕТОНОВ C ДИМЕКАРБИНОМ –НОВЫЙ ПОДХОД К 6-ФТОРАЛКИЛМОДИФИКАЦИИ 5-ГИДРОКСИИНДОЛОВ
В.И. Дяченко, С.М. Игумнов
Институт элементоорганических соединений им. А. Н. Несмеянова РАН, Российская Федерация, 119991, Москва, В-334, ул. Вавилова, д. 28
e-mail: vic-d.60@mail.ru
Аннотация: Гексафторацетон 2а, нитропентафторацетон 2b и перфторциклогексанон 2с гладко реагируют с димекарбином 1 с образованием продуктов С-6-оксиалкилирования 5а-с с выходами 74-90%. В случае метиловых эфиров трифтор- 3 и дифторхлорпировиноградной кислоты 4 реакция оксиалкилирования сопровождается циклизацией промежуточно образующихся эфиров 6а, 7а в соответствующие лактоны 6b, 7b.
Ключевые слова: cеротонин, 5-оксииндолы, гексафторацетон, нитропентафторацетон, метилтрифторпируват, метилдифторхлорпируват, перфторциклогексанон, димекарбин, С-оксиалкилирование.
Производные 5-оксииндола (5-ОИ) занимают особое место в физиологии высшей нервной системы. Серотонин играет важную роль нейротрансмиттера ЦНС человека, регулируя процессы передачи импульсов в нейронах сердечно-сосудистой, эндокринной и других важных систем организма [1,2]. Близкий ему по строению гормон мелатонин участвует в контроле смены дневного и ночного ритмов живых организмов [3]. Синтетические производные 5-ОИ – индометацин и димекарбин на протяжении многих лет используются в медицинской практике как противовоспалительное и гипотензивное средство соответственно [4]. Отечественный препарат арбидол прочно занял позиции на рынке противовирусных препаратов [5,6].
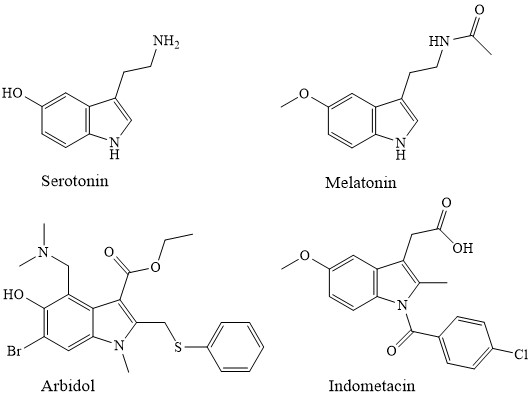
5-ОИ, являясь, с одной стороны, индолами, а с другой - фенолами, имеют богатую химию. Большой вклад в ее развитие внесено Гриневым и сотрудниками. Наиболее характерными для них являются реакции электрофильного замещения: алкилирование, ацилирование, галогенирование, диазотирование, различные варианты аминометилирования [7].
Установлено, что наличие фтора в биологически активных соединениях существенно влияет на их метаболизм [8]. Наличие в молекуле атомов фтора или полифторалкильных групп повышает липофильность фторсодержащих соединений, что облегчает их проникновение через белково-липидную мембрану клетки. Сравнение по объему атома фтора с атомом водорода в органических соединениях, а также по нуклеофильности с атомом кислорода легло в основу, так называемого, «эффекта маскировки» [9]. В связи с этим в настоящее время все больше возрастает интерес к разработке новых методов синтеза фторсодержащих соединений, в частности CF3-cодержащих индолов, структурный скелет которых лежит в основе изучаемых в настоящей работе соединений [10]. Несмотря на значительные успехи в синтетической органической химии, методов одностадийного введения фторсодержащих заместителей в 5‑ОИ в литературе не описано.
Изучив закономерности реакций полифторкарбонильных соединений с фенолами, нафтолами, полифенолами, фенолятами, 8-оксихинолинами [11], мы, на примере взаимодействия полифторкетонов с этиловым эфиром 5-окси-1,2-диметил-1Н-индол-3-ил карбоновой кислоты 1 – димекарбом [4], нашли удобный способ одностадийной фторалкилмодификации 5-ОИ. В данных превращениях, в качестве электрофильных агентов были использованы гексафторацетон 2a, нитропентафторацетон 2b, перфторциклогексанон 2c, метиловый эфир трифторпировиноградной кислоты 3, а также описанный относительно недавно метилдифторхлорпируват 4 [12].
Показано, что нагревание при 120°С на протяжении 1 часа в ледяной уксусной кислоте эквимольных количеств 1 и гексафторацетона приводит к образованию с высоким выходом продукта С-оксиалкилирования 5а (см.Схему 1).
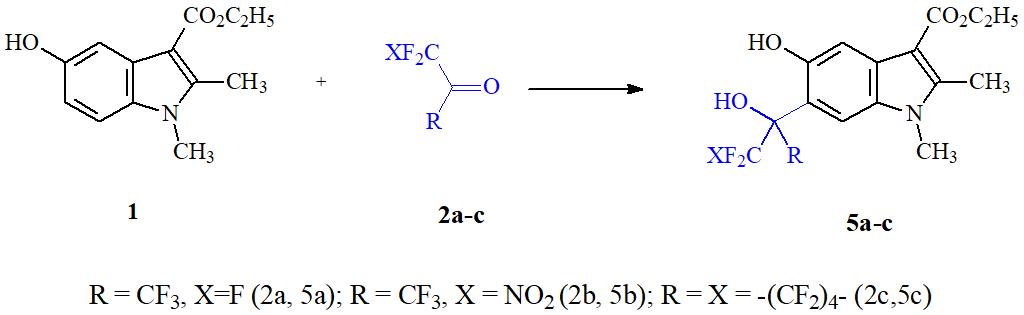
Схема 1.
Реакция осуществляется региоселективно в положение 6 оксииндола 1 с полной конверсией последнего. В этих же условиях взаимодействует c соединением 1 нитропентафторацетон 2b, образуя продукт 5b с выходом 80%. Аналогично гексафторацетону вступает в реакцию с димекарбином и малоизученный в реакция С-оксиалкилирования 2,2,3,3,4,4,5,5,6,6-додекафторциклогексанон (2c), образуя высокофторированный оксииндол (5b) с выходом 74%.
В спектре ЯМР 1Н вновь синтезированных cоединений 5a-с наблюдается значительное уширение сигналов протонов фенольной и полифторизопропанольной ОН-групп. По‑видимому, это связано с интенсивным протонным обменом между их атомами кислорода.
В аналогичных условиях осуществляется взаимодействие с димекарбина с кетоэфирами 3 и 4 (см. схему 2).
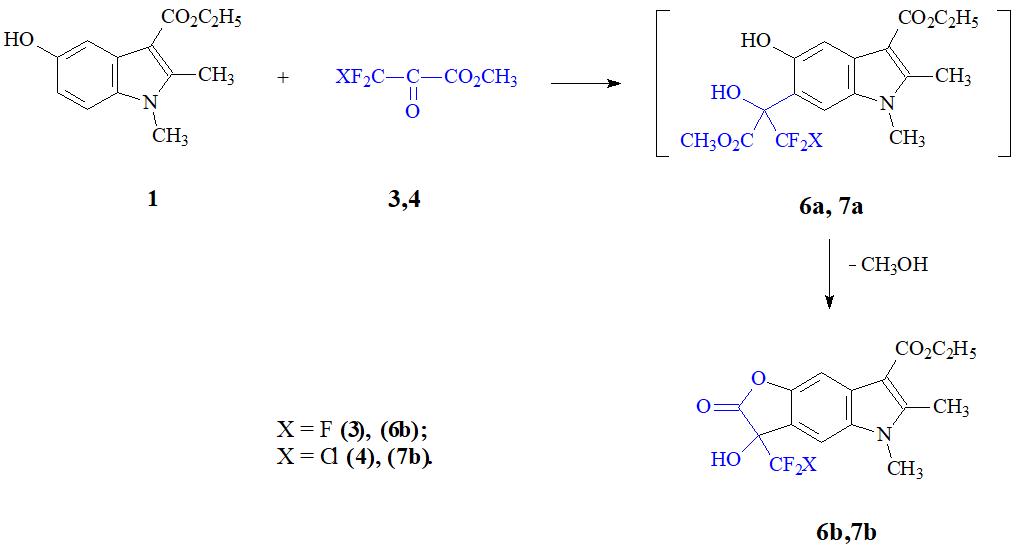
Схема 2.
В этих условиях промежуточно образующиеся продукты С-6-оксиалкилирования 6а, 7а циклизуются в соответствующие лактоны 6b, 7b c выходом 86,7% и 75% соответственно. Следует отметить, что это первый пример использования кетоэфира 4 в реакции С-оксиалкилировании гетароматических соединений подобного типа.
Димекарбин имеет плохую растворимость, и уксусная кислота была нами использована в качестве подходящего высококипящего полярного растворителя. Относительно высокая температура реакции необходима скорее всего для лучшей растворимости исходного 5-ОИ нежели для активации реагентов. По-видимому, С-оксиалкилирование 1 может быть осуществлено и при значительно более низкой температуре.
Таким образом, на примере димекарбина показано, что полифторкарбонильные соединения 2a-с и кетоэфиры 3,4 являются удобными реагентами для одностадийного введения альфа-гидроксиполифторалкильного заместителя в 6 положение 5-ОИ, включая соединения, заведомо обладающие биологической активностью (антибактериальной, противовирусной, гипотензивной и т. п.).
По-видимому, данный метод фторалкилмодификации может быть применим и к другим многочисленным продуктам реакции Неницеску, включая аннелированные оксииндолы более сложного строения.
Наличие в соединениях 2a-с хелатной функции позволяет получать различного рода аннелированые 1,3-диоксафосфоринаны [13], что существенно расширяет синтетические возможности на основе полученных фторсодержащих оксииндолов 5a-с.
Экспериментальная часть
Спектры ЯМР 1Н и 19F полученных соединений 5а, 5с, 6b и 7b сняты в ДМСО-d6 и CDCl3 на приборе Bruker Avance 300 (300 и 282 МГц соответственно), соединения 5b на Bruker Avance 400 (400 и 376 МГц соответственно). Химические сдвиги в ЯМР 1Н спектрах приведены в шкале δ (м.д.) относительно ТМС (внутренний стандарт), в спектрах ЯМР 19F относительно CCl3F (внешний стандарт). Константы спин-спинового взаимодействия приведены в Гц. Элементный анализ выполнен в лаборатории микроанализа ИНЭОС РАН. Контроль протекания реакций осуществляли методом ТСХ на пластинах фирмы «Merck» (силикагель 60 F254, 0.25 мм). Значения RF синтезированных соединений определены в системе ацетон-СCl4 = 1:2.
Этиловый эфир 5-гидрокси-1,2-диметил-6-(2,2,2-трифтор-1-гидрокси-1-трифторметилэтил)-1Н-индол-3-ил карбоновой кислоты (5а)
В стеклянную ампулу помещали 2,33 г (10 ммоль) 1 и 6 мл ледяной уксусной кислоты. Затем ампулу охлаждали до -78°С и конденсировали в нее 2,00 г (12 ммоль) гексафторацетона 2, запаивали и нагревали на масляной бане 1 ч при 120°С. Ампулу охлаждали до -78°С, вскрывали, кристаллический осадок отфильтровывали, промывали уксусной кислотой, затем бензолом и сушили в вакууме. Получали 3,60 г соединения 5а в виде бесцветных кристаллов, выход 90,2%, т.пл. 252-254°С (уксусная к-та), Rf= 0,6 (ацетон-CCl4 = 1:2).
Спектр ЯМР 1Н (ДМСО-d6, δ, м.д., J/Гц): 10,57 (уш.с, 1Н, ОН); 8,83 (уш.с, 1Н, ОН); 7,56 (с, 1Н, Ar); 7,54 (с, 1Н, Ar); 4,27 (кв, 2Н, OCH2CH3, 3JН-Н=7); 3,68 (c, 3Н, NCH3), 2,70 (c, 3Н, CH3), 1,36 (т, 3Н, OCH2CH3, 3JН-Н=7).
Спектр ЯМР 19F (ДМСО-d6, δ, м.д., J/Гц): -73,48.
Найдено (%): С, 47,86; Н, 3,76, N, 3,96. С16H15F6NO4.
Вычислено (%): С, 48,13; Н, 3,79, F 3,51.
Этиловый эфир 6-[1-дифторнитрометил)-2,2,2-трифтор-1-гидроксиэтил]-5-гидрокси-1,2-диметил-1Н-индол-3-ил карбоновой кислоты (5b)
В стеклянную ампулу помещали 1,16 г (5 ммоль) 1, 4 мл ледяной уксусной кислоты и 1,0 г (5,5 ммоль) нитропентафторацетона. Ампулу охлаждали до -78°С запаивали и нагревали на масляной бане 1 ч при 120°С. После охлаждения до -78°С, ампулу вскрывали, ее содержимое переносили в колбу и упаривали на роторном испарителе. Твердый остаток кристаллизовали из нитрометана. получали 1.7 г белого кристаллического соединения 5b,. выход 80%, т.пл. 235°С (субл.) (нитрометан), Rf= 0,51 (ацетон- CCl4 = 1:2).
Спектр ЯМР 1Н (СDСl3, δ, м.д., J/Гц): 10,15 (уш.с, 1Н, ОН); 8,87 (уш.с, 1Н, ОН); 7,13 (с, 1Н, Ar); 7,43 (с, 1Н, Ar); 4,37 (кв, 2Н, OCH2CH3, 3JН-Н=7); 3,73 (c, 3Н, NCH3), 2,74 (c, 3Н, CH3), 1,46 (т, 3Н, OCH2CH3, 3JН-Н=7).
Спектр ЯМР 19F (СDСl3, δ, м.д., J/Гц): -73,66 (т, 3F, CF3, 3JF-F=5,6); -90,17 (кв.кв, 1F, CF2NO2, 2JF-F=165, 3JF-F=7,5); -94,16 (уш.кв.кв, 1F, CF2NO2, 2JF-F=165, 3JF-F=7,5).
Найдено (%): С, 44,80; Н, 3,64; N, 6,49. С16H15F5N2O6.
Вычислено (%): С, 45,08; Н, 3,55; N 6,57.
Этиловый эфир 6-(2,2,3,3,4,4,5,5,6,6-декафтор-1-гидроксициклогексил)-5-гидрокси-1,2-диметил-1Н-индол-3-карбоновой кислоты (5с)
В стеклянную ампулу помещали 1,16 г (5 ммоль) 1, 4 мл ледяной уксусной кислоты и 1,5 г (5,4 ммоль) перфторциклогексанона. Затем ее охлаждали до -78°С запаивали и нагревали на масляной бане 1 ч при 120°С. После охлаждения до -78°С, ампулу вскрывали, ее содержимое упаривали на роторном испарителе, твердый остаток кристаллизовали из нитрометана. Получали 1.9 г белого кристаллического соединения 5с, выход 74%, т.пл. 270°С (субл.) (нитрометан), Rf= 0,56 (ацетон- CCl4 = 1:2).
Спектр ЯМР 1Н (ДМСО-d6, δ, м.д., J/Гц): 11,51 (уш.с, 1Н, ОН); 9,65 (уш.с, 1Н, ОН); 7,66 (с, 1Н, Ar); 7,57 (с, 1Н, Ar); 4,28 (кв, 2Н, OCH2CH3, 3JН-Н=7); 3,67 (c, 3Н, NCH3); 2,70 (c, 3Н, CH3); 1,36 (т, 3Н, OCH2CH3, 3JН-Н=7).
Спектр ЯМР 19F (ДМСО-d6, δ, м.д., J/Гц): - 34,85 (д.д, 2F, CF2, 2JF-F=282); 38,99 (д.д, 2F, CF2, 2JF-F=271); - 42,58 (д.д, 1F, CF2, 2JF-F=267); - 54,44 (д.д, 2F, CF2, 2JF-F=282); 58,28 (д.д, 2F, CF2, 2JF-F=271); 62,50 (д.д, 1F, CF2, 2JF-F=282).
Найдено (%): C, 44,51; Н, 3,15, F 36,81. С19H15F10NO4.
Вычислено (%): C, 44,63; Н, 2,96, F 37,16.
Этиловый эфир 3-гидрокси-5,6-диметил-2-оксо-3-трифторметил-3,5-дигидро-2Н-1-окса-5-аза-s-индацен-7-ил карбоновой кислоты (6b)
В стеклянную колбу, снабженную обратным холодильником с хлоркальциевой трубкой и магнитной мешалкой с нагревом, помещали 2,33 г (10 ммоль) 1, 6 мл ледяной уксусной кислоты, 1,8 г (12 ммоль) 3 и кипятили 1 ч при 120°С. Затем реакционную массу охлаждали до 20°С, осадок отфильтровывали, промывали уксусной кислотой, затем бензолом и сушили в вакууме. Получали 3,1 г cоединения (6b) в виде бесцветных кристаллов,.выход 86,8%, Rf= 0,48 (ацетон-CCl4= 1:2), т.пл. 250-252°С (этанол).
Спектр ЯМР 1Н (ДМСО-d6, δ, м.д., J/Гц): 8,33 (c, 1 Н, ОH); 7,83 (уш.c, 1 Н, Ar); 7,76 (c, 1 Н, Ar); 4,30 (кв, 2 Н, ОСН2СН3, 3Jн-н = 7,0); 3,79 (с, 3 Н, NСН3); 2,74 (с, 3 Н, СН3); 1,36 (т, 3 Н, ОСН2СН3, 3Jн-н = 7,0);
Спектр ЯМР 19F (ДМСО-d6, δ, м.д.): -77,5 (с, 3F, СF3).
Найдено (%): С, 53,83; Н, 4,08; N, 3,96. С16H14F3NO5.
Вычислено (%): С, 53,79; Н, 3,95, N 3,92.
Этиловый эфир 3-(дифторхлорметил)-3-гидрокси-5,6-диметил-2-оксо-3-трифторметил-3,5-дигидро-2Н-окса-5-аза-s-индацен-7-ил карбоновой кислоты (7b)
В стеклянную колбу, снабженную обратным холодильником с хлоркальциевой трубкой и магнитной мешалкой с нагревом помещали 1,17 г (5 ммоль) 1, 3 мл ледяной уксусной кислоты, 1 г (5,8 ммоль) 3 и кипятили 4 ч. Затем реакционную массу упаривали на роторном испарителе, полученный остаток кристаллизовали из нитрометана. Получали 1.4 г белого вещества 7b, выход 75%, Rf= 0,49 (ацетон- CCl4= 1:2), т.пл. 247-248°С (нитрометан).
Спектр ЯМР 1Н (ДМСО-d6, δ, м.д., J/Гц): 8,41 (c, 1 Н, ОH); 7,81 (уш.c, 1 Н, Ar); 7,75 (c, 1 Н, Ar); 4,30 (кв, 2 Н, ОСН2СН3, 3Jн-н = 7,0); 3,78 (с, 3 Н, NСН3); 2,74 (с, 3 Н, СН3); 1,35 (т, 3 Н, ОСН2СН3, 3Jн-н=7,0);
Спектр ЯМР 19F (ДМСО-d6, δ, м.д.): -66,53 (д, 1F, СF2Cl, -65,97 (д, 1F, СF2Cl, 2JF-F=163,8);
Найдено (%): С, 51,27; Н, 3,87; N, 3,88. С16H14СlF2NO5.
Вычислено (%): C, 51,42; Н 3,78; N, 3,75.
Благодарности
Работа выполнена в рамках Государственного задания № 075-03-2023-642 Министерства науки и высшего образования Российской Федерации. ЯМР спектральные исследования синтезированных соединений проведены при поддержке Российского научного фонда (грант № 22-23-00559) с использованием научного оборудования Центра исследования строения молекул ИНЭОС РАН.
Авторы выражают благодарность д.х.н. Осипову С.Н. за представленный для исследований метиловый эфир дифторхлорпировиноградной кислоты.
Список литературы
- S. N. Young, J. Psychiatry Neurosci, 2007, 32(6), 394-399.
- S. Benmansour, M. Cecchi, D. A. Morilak, G. A. Gerhardt, M. A. Javors, G. G. Gould, A. Frazer, J. of Neuroscience, 1, 1999, 19(23), 10494-10501.
- L. A. Newman, M. T. Walker, R. L. Brown, T. W. Cronin, P. R. Robinson, Biochemistry, 2003, 42(44), 12734-12738.
- M. D. Mashkovsky, Medicines (a manual on pharmacotherapy for doctors), 1978, Ed. Medicine, 1344 p.
- WO9008135 (1990).
- Ф. А. Трофимов, Н. Г. Тишкова, С. А. Зотова, А. Н. Гринев, Хим.-фарм. журн., 1993, 1, 70‑71.
- (a) Grinev, A. N. et al., Pharmaceutical Chemistry Journal, 1970; 1, 25-29; (b) Kurilo G. N.; Rostova N. I.; Cherkasova A. A., Turchin, K. F, Alekseeva L. M., Grinev A. N., Chemistry of Heterocyclic Compounds, 1980, 1043–1047; (c) Trofimov F. A., Tsyshkova N. G., Zotova S. A., Grinev A. N., Pharmaceutical Chemistry Journal, 1993, 27, 1, 75-76.
- M. Novak, K. J. Kayser, M. E. Brooks, J. Org. Chem., 1998, 63, 5489-5496.
- Ishikawa N., Fluorine compounds. Synthesis and application, M.: Mir. 1990. 407 p.
- 10 (a) V. M. Muzalevskiy, Z. A. Sizova, V. G. Nenajdenko, Molecules, 2021, 26(16), 5084; (b) D. V. Vorobyeva, T P. Vasilyeva, S. N. Osipov, Russ. Chem. Bull., 2022, 71, 1949; (c) Sigan A. L., Volkonskii A. Yu, Kagramanov N. D., Guseva E. V., Chkanikov N. D., Fluorine notes, 2023, 4(149), 1-2.
- (a) Dyachenko V. I., Galakhov M. V., Kolomiets A. F., Fokin A. V., Bull. Acad. Sci. USSR, Div. Chem. Sci (Engl. Transl.), 1989, 38, 4.2, 831-836; (b) Dyachenko, V. I., Kolomiets, A. F., Fokin A. V., Bull. Acad. Sci. USSR, Div. Chem. Sci (Engl. Transl.), 1987, 36, 995-1000; (c) Dyachenko V. I., Kolomets A. F., Fokin, A. V., Bull. Acad. Sci. USSR, Div. Chem. Sci (Engl. Transl.), 1987, 36, 12, 2646-2648; (d) Dyachenko V. I., Galakhov M. V., Kolomiets A. F., Fokin A. V., Bull. Acad. Sci. USSR, Div. Chem. Sci (Engl. Transl.), 1989, 38, 12, 2550-2553;
- S. N. Osipov, A. S. Golubev, N. Sewald, T. Michel, A. F. Kolomiets, A. V. Fokin, K. Burger., J. Org. Chem., 1996, 61, 21, 7521–7528.
- (a) E. E. Nifant’ev, T. S. Kukhareva, V. I. Dyachenko, A. F. Kolomiets, N. S. Magomedova, V. K. Bel’skii and L. K. Vasyanina, Phosphorus Sulfur Silicon Relat. Elem., 1994, 92, 29-38; (b) V. I. Dyachenko, A. A. Korlyukov, Mendeleev Commun., 2019, 29, 89–90.
Статья рекомендована к публикации к.х.н. О.В. Брызгаловой
Fluorine Notes, 2023, 150, 5-6


