Поступило в редакцию: Сентябрь 2023
УДК 547.46'054.74+547.582.4+547.826.7+547.46'054.72+547.562
Fluorine Notes, 2023, 150, 3-4
СИНТЕЗ ФТОР- ИЛИ ХЛОРСОДЕРЖАЩИХ п-ПИРИДИНИЛОКСИЗАМЕЩЕННЫХ БЕНЗАМИДОКСИМОВ
Т. П. Васильева*, Д. В. Воробьева
Институт элементоорганических соединений им. А.Н. Несмеянова РАН, Российская Федерация, 119334 Москва, ул. Вавилова, 28, стр. 1
Факс: (499) 135 5085. E-mail: d-20@mail.ru
Аннотация: Модельной реакцией трихлорпиридина 1 с 4-цианофенолом в присутствии K2CO3 в ДМФА получен новый 4-((3,5-дихлорпиридин-4-ил)окси)бензонитрил 2. В аналогичных условиях действием 4-цианофенола на 2-хлор-5-(трифторметил)пиридин синтезирован ранее труднодоступный 4-((5-трифторметил)пиридин-2-ил)-окси)бензонитрил 4 с высоким выходом. На основе полученных нитрилов 2 и 4 разработаны препаративные методы синтеза новых фтор- или хлорсодержащих п-пиридинилоксизамещенных бензамидоксимов 3 и 5, пригодные для масштабирования.
Ключевые слова: 3,4,5-трихлорпиридин, 4-цианофенол, 2-хлор-5-(трифторметил)пиридин, фтор- или хлорсодержащие п-пиридилоксизамещенные бензонитрилы и бензамидоксимы, гидроксиламин.
Ранее нами был разработан метод получения фторзамещенных бензамидов [1]. Данная работа посвящена другим производным – п-пиридилоксизамещенным бензонитрилам и амидоксимам. Известно, что ключевые фрагменты диариловых эфиров (в том числе и содержащих пиридиновое ядро) фигурируют в некоторых инсектицидах (пирипроксифен, феноксикарб) и гербицидах (фюзилад, хлометоксинил) [2]. В отличие от бензонитрилов бензамидоксимы (N-гидрокси-бензимидамиды) – малоизученный класс соединений с потенциальной биологической активностью. Так, сообщалось о высокой антималярийной активности амидоксима 3 [3], однако в работе нет методики его синтеза и физических характеристик. Мы предположили, что удобным исходным соединением для получения как вещества 3, так и соответствующего нитрила 2 мог бы явиться легкодоступный 3,4,5-трихлорпиридин 1 (рисунок 1).
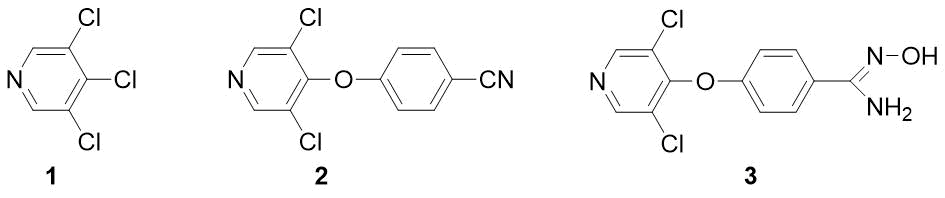
Рисунок 1.
Целью настоящей работы является разработка препаративных синтезов соединений 2 и 3 и использование этих методик для получения CF3-содержащих аналогов (нитрила и амидоксима). Известно, что фторсодержащие лекарства обладают повышенной активностью по сравнению с гидрированными аналогами [4,5]. До наших работ 3- или 4‑пиридилоксизамещенные бензонитрилы получали реакцией 4- или 2-фторпиридинов с цианофенолами в среде ДМФА в присутствии Et3N [2] или карбоната калия [6] (схема 1).
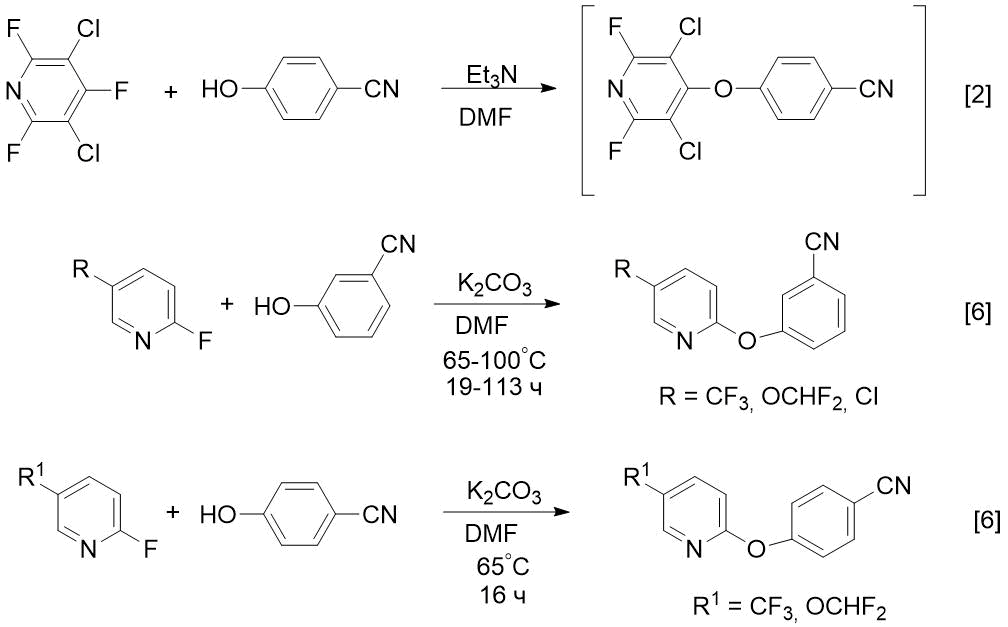
Схема 1. Известные способы получения м- или п-пиридилоксизамещенных бензонитрилов из 4- или 2-фторпиридинов [2,6].
Недостатком этих способов является высокая стоимость фторпиридинов (например, 2-фтор-5-трифторметилпиридина). Нами разработан более экономичный метод синтеза подобных нитрилов на основе доступных хлорпиридинов, в частности, трихлорпиридина 1. Ранее 1 получали с низким выходом (наряду с другими продуктами) реакциями пиридина [7] или α-пиколина [8] с хлором. Известно также о синтезе 1 взаимодействием PCl5 и POCl3 с 3,5-дихлор-4-пиридоном (без указания выхода) [9], однако в этом случае из-за агрессивного реактива (PCl5) и высокой температуры (125°С) происходило осмоление реакционной смеси. По нашим данным, оптимальным методом получения 1 является взаимодействие 3,5-дихлор-4-пиридона с двойным избытком POCl3 в присутствии каталитических количеств диметилформамида и умеренном нагревании. Реакцией полученнного таким способом трихлорпиридина 1 с 4-цианофенолом в присутствии K2CO3 нам удалось получить целевой нитрил 2 с высоким выходом, при этом происходило замещение атома хлора в положении 4 пиридинового цикла (схема 2).
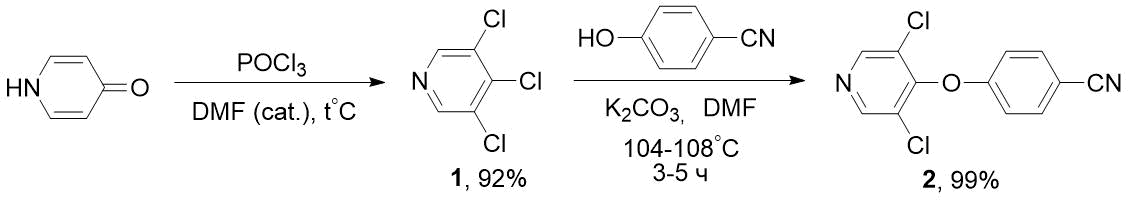
Схема 2. Синтез 4-((3,5-дихлорпиридин-4-ил)окси)-бензонитрила 2 из 3,4,5-трихлорпиридина 1.
Для получения F-содержащего пиридилоксибензонитрила 4 в качестве исходного соединения мы использовали 2-хлор-5-(трифторметил)пиридин, который значительно дешевле 2-фтор – аналога. Синтез проводили аналогично получению нитрила 2, но в более жестких условиях – при 103-128°С в течение 8-10 ч (схема 3). Тем не менее 2-Cl-5-CF3-пиридин оказался более реакционноспособным, чем 2-F – аналог, для которого время реакции составляло 16 ч [6] (см. схему 1).
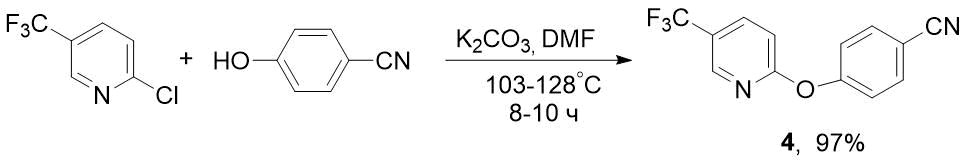
Схема 3. Синтез 4-((5-(трифторметил)пиридин-2-ил)окси)бензонитрила 4 из 2-хлор-5-(трифторметил)пиридина.
Пиридилоксизамещенные бензамидоксимы ранее получали нагреванием смеси соответствующих нитрилов, хлоргидрата гидроксиламина и бикарбоната натрия в водно-спиртовой среде, но образующиеся целевые продукты использовались в синтезе лекарственных средств без очистки [6] (схема 4).
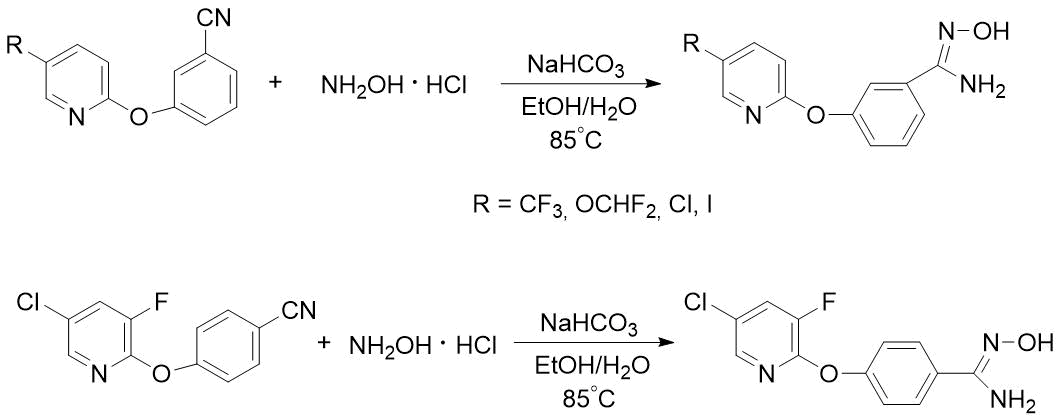
Схема 4. Описанные в литературе синтезы м- или п-пиридилоксизамещенных бензамидоксимов [6].
На примере получения гетероциклических амидоксимов 3 и 5 нами показано, что более чистые продукты образуются при проведении процесса в два этапа: 1) генерирование in situ гидроксиламина действием углекислого натрия на NH2OH·HCl в воде; 2) прибавление раствора NH2OH к нитрилам 2 или 4 в спирте с последующим нагреванием (схема 5).
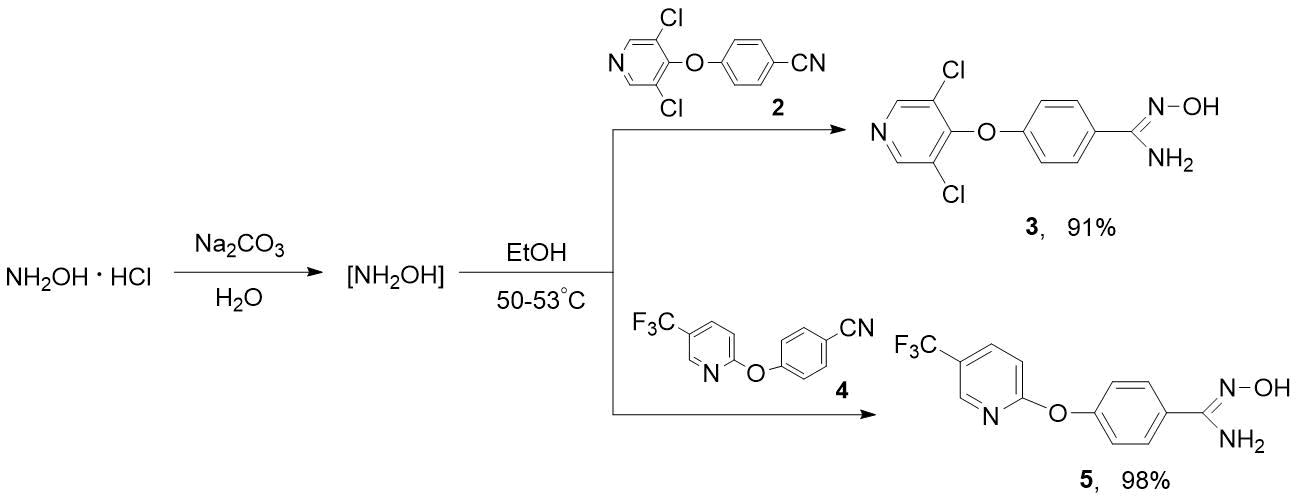
Схема 5. Синтез гетероциклических амидоксимов 3 и 5.
Очевидно, что разработанный подход к синтезу соединений 3 и 5 может быть успешно применен для получения и других полифтор- и полифторалкилсодержащих амидоксимов. Синтезированные амидоксимы 3 и 5 – твердые кристаллические вещества, которые могут быть очищены переосаждением из раствора в водном бикарбонате натрия добавлением 2н соляной кислоты. Состав и строение этих соединений доказаны данными спектроскопии ЯМР и элементного анализа. В спектрах 1Н ЯМР 3, 5 характерными являются синглеты гидроксильных протонов (=N-OH) при 9,62-9,67 м.д. и аминогрупп (NH2) в области 5,80-5,86 м.д. Сигнал CF3-группы амидоксима 5 в спектре 19F ЯМР наблюдается при ‑60,04 м.д. В масс-спектре 5 молекулярный ион является базовым.
Таким образом, в результате настоящей работы на основе 2-хлор-5-(трифторметил)пиридина разработан экономичный синтез ранее труднодоступного нитрила 4 с высоким выходом. Это соединение является важным объектом с фармакофорным ядром и используется в синтезах триазиновых ингибиторов ангиогенеза (для лечения рака и макулодистрофии глаз) [10], гетероарильных производных аминопропанола, применимых при терапии астмы, аллергии и атеросклероза [6], а также производных 1,2,4-оксадиазолов, предложенных для лечения аутоимунных и сосудистых болезней [11]. Что касается синтезированных амидоксимов, то поскольку дихлорзамещенное соединение 3 оказалось биологически активным [3], полученный нами новый CF3-содержащий аналог 5, безусловно, представляет не меньший интерес для медицинской химии, чем исходный нитрил 4. Это подтверждается тем, что некоторые бензамидоксимы, содержащие в п- или м-положении замещенные (пиридин-2-ил)окси-группы, часто используются для получения лекарственных веществ [6].
Экспериментальная часть.
Cпектры ЯМР регистрировали на приборах «Bruker WM-250» и «Bruker Avance-300» с рабочими частотами 250 МГц и 300 МГц (внутренний стандарт SiMe4) для 1Н ЯМР и 282 МГц для 19F ЯМР (CFCl3) соответственно. Элементный анализ выполнен в лаборатории микроанализа ИНЭОС РАН. Контроль протекания реакций методом ТСХ осуществляли на пластинах фирмы «Merck» (силикагель 60 F254, 0.25 мм). Значения RF полученных соединений определены в системах ацетон-СCl4 = 1:10 или этилацетат - петролейный эфир = 2:1. Исходные 3,5-дихлор-4-пиридон, 2-хлор-5-(трифторметил)пиридин и 4-цианофенол являются коммерчески доступными реагентами. Все используемые растворители очищали по стандартным методикам.
3,4,5-Трихлорпиридин (1)
К смеси 46,0 г (0,28 моль) 3,5-дихлор-4-пиридона и 86,3 г (0,56 моль) хлорокиси фосфора прибавляли по каплям 4,2 мл ДМФА. После перемешивания при 20°С в течение 1 часа реакционную смесь кипятили с обратным холодильником до завершения реакции (3‑5 ч), затем выливали в стакан с ледяной водой (650 мл), и смесь нейтрализовали до рН~5-7 прибавлением 40%-ной NaOH (139 мл). Выпавший осадок отфильтровывали и трижды промывали холодной водой, затем холодным водным спиртом (1:1) (80 мл). Продукт сушили на воздухе, затем в вакууме – сначала над CaCl2, затем над P2O5. Получали 47,09 г (92%) соединения 1 в виде порошка светло-бежевого цвета с т. пл. 78-79°С (в [7] 74-75°С, в [8] 73‑74°С, в [9] 76-77°С, RF (раствор в CHCl3) 0,82 (в системе CHCl3 – МеОН = 37:3); 0,68 (в системе ацетон - CCl4 = 1:10)
Спектр 1Н ЯМР (300 МГц, CDCl3, δ, м.д.): 8,53 (с, 2Н, 2СН).
4-((3,5-Дихлорпиридин-4-ил)окси)бензонитрил (2)
К раствору 46,8 г (0,26 моль) трихлорпиридина 1 и 30,52 г (0,26 моль) 4-цианофенола в 210 мл ДМФА прибавляли 42,47 г (0,31 моль) измельченного К2CO3, затем реакционную смесь нагревали при перемешивании при 104-108°С до завершения реакции (3-5 ч). Содержимое колбы охлаждали и выливали в стакан с ледяной водой (1320 мл). Образующийся осадок отфильтровывали, трижды промывали холодной водой, затем сушили на воздухе. Получили 67,0 г (99%) соединения 2 в виде светлых кристаллов с т. пл. 141‑143°С и RF = 0,35 (раствор в CHCl3, в системе ацетон - CCl4 = 1:10).
Спектр 1Н ЯМР (250 МГц, CDCl3, δ, м.д., J/Гц): 6,91 (д, 2Н, Ar, J = 9,0); 7.66 (д, 2Н, Ar, J = 9,0); 8.61 (с, 2Н, 2СНPy).
Найдено (%): С, 54,51; Н, 2,26; N, 10,59. C12H6Cl2N2O.
Вычислено (%): С, 54,34; Н, 2,26; N, 10,57.
4-((3,5-Дихлорпиридин-4-ил)окси)-N'-гидроксибензимидамид (3)
Стадия 1. Получение раствора гидроксиламина в воде. К раствору 26,77 г (0,25 моль) Na2CO3 в 102 мл теплой (~42°С) воды постепенно прибавили 35,14 г (0,5 моль) солянокислого гидроксиламина, и перемешивали при этой температуре до прекращения выделения CO2.
Стадия 2. К суспензии 66,93 г (0,25 моль) нитрила 2 в 1100 мл этанола прибавляли теплый раствор NH2OH (из стадии 1) при 43°С при перемешивании, затем полученную смесь нагревали при 51-53°С до завершения реакции (6-7 ч, ТСХ-контроль в системе этилацетат – петролейный эфир = 2:1). Реакционную смесь охлаждали до 0÷5°С, осадок отфильтровывали и промывали холодными растворителями: сначала водой (65 мл х 3 раза), затем метанолом (55 мл х 2 раза) и петролейным эфиром (45 мл х 2 раза). Продукт сушили на воздухе и в вакууме – сначала над CaCl2, затем над P2O5. Получили 68,46 г (91%) соединения 3 в виде белого порошка с т. пл. 224-225°С и RF (раствор в теплом метаноле) 0,27-0,35 (в системе этилацетат – петролейный эфир = 2:1); 0,59 (в этилацетате).
Спектр 1Н ЯМР (300 МГц, d6-DMSO, δ, м.д., J/Гц): 5,80 (с, 2Н, NH2); 6,93 (д, 2Н, Ar, J = 8,6); 7,66 (д, 2Н, Ar, J = 8,6); 8,79 (с, 2Н, 2СНPy); 9,62 (с, 1Н, =N-OH).
Найдено (%): С, 48,11; Н, 2,87; N, 13,99. C12H9Cl2N3O.
Вычислено (%): С, 48,32; Н, 3,02; N, 14,09.
4-((5-(Трифторметил)пиридин-2-ил)окси)бензонитрил (4)
Синтез проводили аналогично получению нитрила 2 из 89,58 г (0,49 моль) 2-хлор-5-(трифторметил)пиридина, 58,73 г (0,49 моль) 4-цианофенола и 81,73 г (0,59 моль) К2CO3 в 280 мл ДМФА, но при более длительном нагревании (8-10 ч), постепенно повышая температуру бани от 103°С до 128°С – до исчезновения цианофенола (ТСХ-контроль в системе ацетон - CCl4 = 1:10). Продукт промывали водой и холодным петролейным эфиром (100 мл), сушили на воздухе, затем в вакууме над CaCl2. Получили 126,4 г (97%) соединения 4 в виде белых кристаллов с т. пл. 85-87°С (из EtOH) и RF = 0,49 (раствор в CH2Cl2, в системе ацетон - CCl4 = 1:10).
Спектр 1Н ЯМР (ср. лит. [6]) (300 МГц, СDCl3, δ, м.д., J/Гц): 7,11 (д, 1Н, СНPy, J = 7,1); 7.24-7.29 (м, 2Н, Ar); 7,71 (д, 2Н, Ar, J = 8,7); 7,96 (дд, 1Н, СНPy, J = 8,7, J = 1,8); 8,41 (с, 1Н, СНPy).
N'-Гидрокси-4-((5-трифторметил)пиридин-2-ил)окси)бензимидамид (5)
Получали аналогично синтезу амидоксима 3. Раствор гидроксиламина (из 23,65 г, 0,34 моль NH2OH·HCl и 18,04 г, 0,17 моль Na2CO3 в 68 мл воды) прибавляли к раствору 63,7 г (0,24 моль) нитрила 4 в 980 мл этанола при 41-42°С, затем реакционную смесь перемешивали при 50°С в течение 5-6 ч. Температуру смеси доводили до 20°С, выпавший осадок (NaCl) отфильтровывали и промывали метанолом (50 мл х 2 раза). Маточник упаривали в вакууме до объема ~250 мл, остаток охлаждали и смешивали с холодной водой (500 мл). Образующийся осадок отфильтровывали, промывали холодной водой (80 мл по 2 раза) и сушили – сначала на воздухе, затем в вакууме (над CaCl2 и P2O5). Получили 70,2 г (98%) соединения 5 в виде блестящих кристаллов белого цвета с т. пл. 138-141°С (после очистки переосаждением NaHCO3 из раствора в 2н соляной кислоте т.пл. 140-143°С) и RF (раствор в МеОН) 0,45 (в системе этилацетат – петролейный эфир = 2:1).
Спектр 1Н ЯМР (300 МГц, d6-DMSO, δ, м.д., J/Гц): 5,86 (с, 2Н, NH2); 7,21 (д, 2Н, Ar, J = 8,4); 7,27 (д, 1Н, СНPy, J = 7,8); 7,74 (д, 2Н, Ar, J = 8,4); 8,24 (д, 1Н, СНPy, J = 7,8); 8,57 (с, 1Н, СНPy), 9,67 (с, 1Н, =N-OH).
Спектр 19F ЯМР (282 МГц, d6-DMSO, δ, м.д.): -60,04 (с, 3F, СF3).
Масс-спектр, m/z: 297 [M+], 100%.
Найдено (%): С, 52,59; Н, 3,31; N, 14,04. C13H10F3N3O2.
Вычислено (%): С, 52,53; Н, 3,37; N, 14,14.
Благодарности
Работа выполнена в рамках Государственного задания № 075-03-2023-642 Министерства науки и высшего образования Российской Федерации с использованием научного оборудования Центра исследования строения молекул ИНЭОС РАН.
Список литературы
- Vasilyeva T.P., Dyachenko V.I., Vorobyeva D.V., Fluorine notes, 2022, 143(4), 3-4.
- Литвак В.В., Майнагашев И.Я., Буханец О.Г., Изв. АН, Сер. Хим., 2007, 945.
- Rastelli G., Pacchioni S., Sirawaraporn W., Sirawaraporn R., Parenti M., Ferrari A., J. Med. Chem., 2003, 46, 2834.
- Ricci G., Ruzziconi R., J. Org. Chem., 2005, 70, 611.
- Zhu Y., Han J., Wang J., Shibata N., Sodeoka M., Soloshonok V.A., Coelho J., Toste F., Chem. Rev., 2018, 118, 3887.
- Kordikowski A., Liu Y., Lüӧnd R., Markert C., Miltz W., Roehn T., Spanka C., Thoma G., Xie T., WO 2022/219546A1, 2022.
- Wibaut J., Nicolai J., Rec. Trav. Chem. Pays-Bas, 1939, 58, 709.
- Шленкова Е.К., Куприянова Н.С., Буткова О.Л., Кацобашвили В.Я., Беэр А.А., Журн. орган. химии, 1977, 13, 446.
- Dohrn M., Diedrich P., Justus Liebigs Ann. Chem., 1932, 494, 284.
- Henkin J., Davidson D., Sheppard G., Woods K., McCroskey R., WO 99/31088, 1999.
- DAS, Jagabandhu, KO, Soo Sung, WO 2012/040532, 2012.
Статья рекомендована к публикации членом редколлегии к.х.н. М.А. Манаенковой
Fluorine Notes, 2023, 150, 3-4


