Поступило в редакцию: Декабрь 2022
УДК 547.412.91; 542.9
Fluorine Notes, 2022, 140, 7-8
ДЕКАРБОКСИЛИРОВАНИЕ 2,3,3,3-ТЕТРАФТОР-2-ИОДПРОПИОНОВОЙ КИСЛОТЫ, ЕЁ КАЛИЕВОЙ СОЛИ И ТРИМЕТИЛСИЛИЛОВОГО ЭФИРА
В.Э. Бойкоaб, A. В. Едуновб, С.М. Игумновaб
аИнститут элементоорганических соединений им. А. Н. Несмеянова РАН,
119991, Москва, ул. Вавилова, д. 28.
e-mail: boykii@mail.ru
бООО НПО «ПиМ-Инвест», 119991, Москва, Ленинский проспект, 47.
Аннотация: Изучено получение 1,1,1,2-тетрафтор-2-иодэтана различными способами, показано, что наилучшим методом является способ, при котором происходит постепенное добавление раствора кислоты в диметилформамиде в заранее разогретый до 140С диметилформамид, предложен механизм данной реакции с образованием CF3CFI – аниона.
Ключевые слова: иодфторалканы, декарбоксилирование, 2,3,3,3-тетрафтор-2-иод-пропионовая кислота.
Иодфторалканы являются базовыми синтонами фторорганической химии, они широко используются для фторалкилирования алифатических, ароматических, гетероциклических, элементоорганических соединений.[1-3]. Низшие иодфторалканы применяются также как телогены при теломеризации с образованием продуктов, используемых для синтеза фторокислот [4], фторированных спиртов [5]; мономеров для термо- и химически стабильных полимеров [6]
Недавно нами был предложен оригинальный метод получения пентафторэтилиодида из перфторпропилена [7], ключевым интермедиатом в котором является фторангидрид 2,3,3,3-тетрафтор-2-иодпропионовой кислоты I. В продолжение этой работы было предложено получить 1,1,1,2-тетрафтор-2-иодэтан и 1,1,2-трифтор-2-иодэтен, используя это же промежуточное соединение.
Известно получение 1,1,1,2-тетрафтор-2-иодэтана декарбоксилированием солей тетрафторпропионовой кислоты в присутствии иода в диметилформамиде с выходом около 30% [8], присоединением HF к 1-иод-1,2,2,2-тетрафторэтану в газовой фазе на хромовом катализаторе [9], иодфторированием 1,1,2-трифторэтена смесью иода и JF5 с использованием алюминия с иодистым алюминием в качестве катализаторов при температурах 130-140С в автоклаве [10], а также взаимодействием 1,1,2-трифторэтена с ICl в среде HF/BF3 при 25С с выходом 73% [11].
Целью данной работы являлось получение 1,1,1,2-тетрафтор-2-иодэтана и 1,1,2-трифтор-2-иодэтена из 2,3,3,3-тетрафтор-2-иодпропионовой кислоты II и её триметилсилилового эфира III, которые, в свою очередь, были получены из фторангидрида I по приведенной ниже Схеме 1.
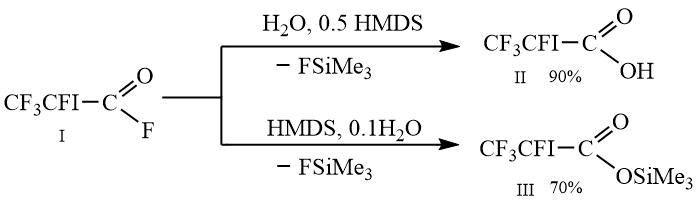
Схема 1.
В зависимости от условий проведения декарбоксилирования кислоты II (Схема 2) нами были получены различные результаты. При проведении процесса декарбоксилирования с одновременной отгонкой образующихся летучих продуктов выход 1,1,1,2-тетрафтор-2-иодэтана I составил 48% (Способ 1). В случае использования метода, заключающегося в постепенном добавлении раствора кислоты в диметилформамиде в заранее разогретый до 140С диметилформамид (Способ 2) удалось увеличить выход желаемого продукта IV до 72%. Однако проведение реакции без одновременной отгонки летучих продуктов (Способ 3) привело к неожиданному результату. После выделения и сопоставления данных ГХ, спектров ЯМР 1Н, 19F и масс-спектров оказалось, что продуктами реакции помимо IV являются 1,1,1,2-тетрафтор-2,2-дииодэтан V и 2,3,3,3-тетрафторпропионовая кислота VI.
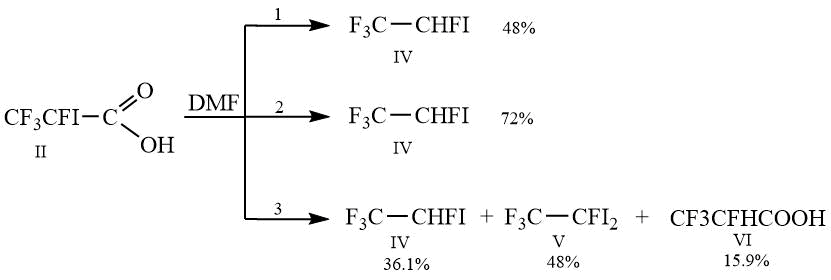
Схема 2.
Декарбоксилирование силилового эфира III не привело к образованию желаемого 1,1,2-трифтор-2-иодэтена VII и триметил(1,2,2,2-тетрафтор-1-иодэтил)силана IX, единственным продуктом реакции оказался 1,1,1,2-тетрафтор-2,2-дииодэтан V.
Как альтернативный вариант получения 1,1,2-трифтор-2-иодэтена VIII было проведено декарбоксилирование калиевой соли 2,3,3,3-тетрафтор-2-иодпропионовой кислоты, однако и в этом случае не произошло образование ожидаемых продуктов, результатом реакции стали соединения IV и V в соотношении 1:3.
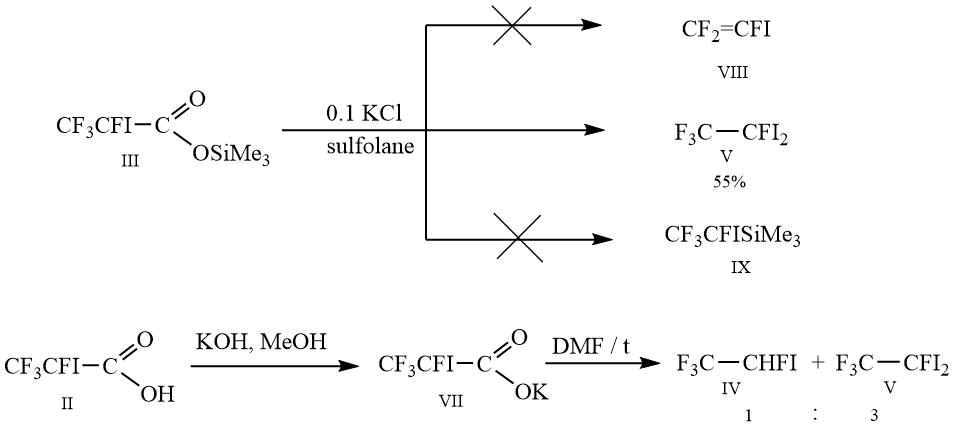
Схема 3.
Анализируя полученные экспериментальные данные, можно предположить, что в результате декарбоксилирования образуется устойчивый CF3CFI – анион, который вступает во взаимодействия, представленные на Схемах 4 и 5.
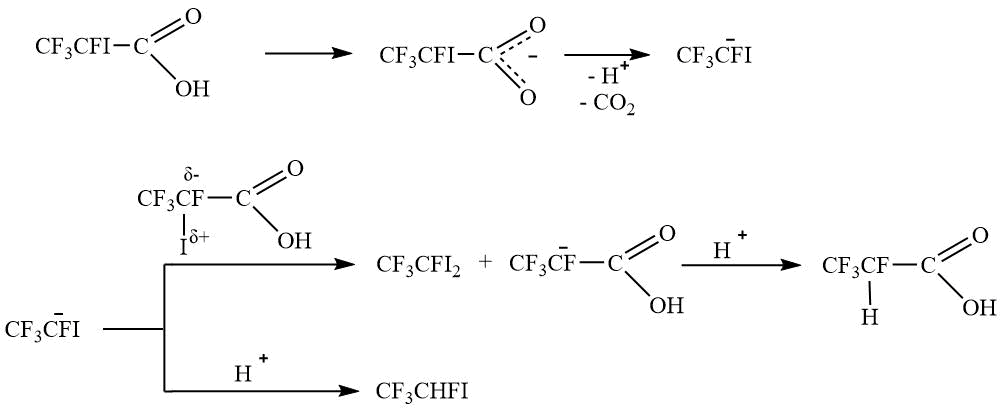
Схема 4.
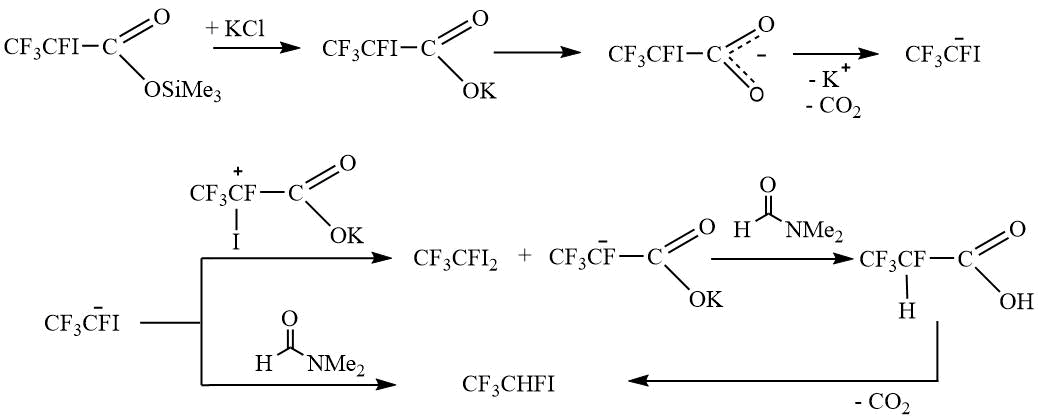
Схема 5.
Таким образом, преимущественно происходит образование дииодпроизводного V, а не предполагаемых алкена VIII или триметилсилана IX.
Экспериментальная часть
1H, 19F ЯМР спектры записаны на спектрометре “Bruker AVANCE-300” при 300 и 282 MHz соответственно, внешний стандарт CDCl3. Химические сдвиги для 1H ЯМР спектров приведены в м.д. относительно ТМС. Химические сдвиги спектров 19F ЯМР приведены в м.д. относительно CFCl3.
Фторангидрид I был получен, как описано в [7].
Получение 2,3,3,3-тетрафтор-2-иодпропионовой кислоты II
К 6.9 г (0.383 моль) воды и 31.1 г (0.192 моль) ГМДС при перемешивании добавляют по каплям 105 г (0.383 моль) фторангидрида I. По мере добавления фторангидрида температура растет до 34С и наблюдается выделение триметилфторсилана. Реакционную массу выдерживают при этой температуре в течение двух часов, затем в вакууме отгоняют в охлаждаемую смесью сухого льда с ацетоном приемную колбу 15 г фтортриметилсилана. Далее приемную колбу заменяют, в куб добавляют 0.2 г порошка меди и отгоняют 94 г 2,3,3,3-тетрафтор-2-иодпропионовой кислоты II в виде бесцветной жидкости. Выход 90%, чистота 97%, Т.пл. 28-31°С, Т.кип. 67-69°С/20 мм рт.ст.
1H ЯМР, δ, м.д: 10.1 (с, 1H, CООН);
19F ЯМР, δ, м.д.: -142.3 (м, 1F, CFI), -76.5 (м, 3F, CF3).
Получение триметилсилилового эфира 2,3,3,3-тетрафтор-2-иодпропионовой кислоты III
К 0.7 г (0.0394 моль) воды и 64 г (0.394 моль) ГМДС при перемешивании добавляют по каплям 108 г (0.394 моль) фторангидрида I. По мере добавления фторангидрида температура растет до 46С и наблюдается выделение триметилфторсилана. Реакционную массу нагревают до 62С и выдерживают при этой температуре в течение 1 часа, затем в вакууме отгоняют в охлаждаемую смесью сухого льда с ацетоном приемную колбу 10 г смеси фтортирметилсилана и ГМДС. Далее приемную колбу заменяют, в куб добавляют 0.2 г порошка меди и отгоняют 95 г триметилсилилового эфира 2,3,3,3-тетрафтор-2-иодпропионовой кислоты III в виде бесцветной жидкости. Выход 70%, чистота 97% по ГХ, Т.кип. 52-53°С/20 мм рт.ст.
1H ЯМР, δ, м.д.: 0.4 (с, 9Н SiMe3);
19F ЯМР, δ, м.д.: -138.6 (м, 1F, CFI), -76.8 (м, 3F, CF3).
Получение 1,1,1,2-тетрафтор-2-иодэтана IV
Способ 1.
Раствор 20 г (0.0735 моль) II в 20 г ДМФА при перемешивании постепенно нагревают до температуры 135-137С, при которой начинается выделение газа, далее реакционную смесь медленно нагревают до 145С, одновременно отгоняя в охлаждаемую смесью сухого льда с ацетоном приемную колбу 1,1,1,2-тетрафтор-2-иодэтана. Выход 48%, чистота 97%, Т.кип. 39°С.
Получение 1,1,1,2-тетрафтор-2-иодэтана IV
Способ 2.
К 20 г ДМФА, нагретого до 140С, при перемешивании добавляют по каплям 20 г (0.0735 моль) II в 15 г ДМФА, одновременно отгоняя 12 г 1,1,1,2-тетрафтор-2-иодэтана в охлаждаемую смесью сухого льда с ацетоном приемную колбу. Выход 72%, чистота 97%, Т.кип. 39°С.
Получение 1,1,1,2-тетрафтор-2-ииододэтана IV
Способ 3.
Раствор 20 г (0.0735 моль) II в 20 г ДМФА помещают в колбу с обратным холодильником, соединенным с холодильником глубокого охлаждения, и при перемешивании постепенно нагревают до 140С. Выдерживают при этой температуре в течение 30 минут до завершения выделения газа, затем реакционную массу охлаждают до комнатной температуры и выливают в двукратный объём 15% соляной кислоты. Нижний слой отделяют и промывают 2x15 мл воды, сушат над CaCl2. Получают 17 г смеси, содержащей по данным ГХ 36.1% IV, 48% V, 15.9% VI.
Получение 1,1,1,2-тетрафтор-2,2-дииодэтана V
Раствор 20 г (0.0579 моль) триметилсилилового эфира III и 0.4 г хлорида калия (0.00579 моль) в 20 г абс. сульфолана при перемешивании постепенно нагревают до температуры 90-93С, при которой начинается выделение газа. Далее реакционную смесь постепенно нагревают до 150°С, одновременно отгоняя в охлаждаемую смесью сухого льда с ацетоном приемную колбу 5 г триметилфторсилана. Реакционную массу охлаждают до комнатной температуры и выливают в двукратный объём воды, промывают 2x15 мл воды, сушат над CaCl2, перегоняют. Получают 6.5 г 1,1,1,2-тетрафтор-2,2-дииодэтана, чистота 96%, Т.кип. 128-130°С.
19F ЯМР, δ, м.д.: -97.7 (м, 1F, CFI2), -80.6 (м, 3F, CF3).
Получение калиевой соли 2,3,3,3-тетрафтор-2-иодпропионовой кислоты VII
К раствору 10 г (0.0367 моль) кислоты II в 15 мл метанола при перемешивании добавляют по каплям 2 г (0.0367 моль) гидроксида калия в 7 мл метанола. Перемешивают 20 минут, метанол отгоняют в вакууме водоструйного насоса, соль сушат над P2O5. Выход количественный.
Декарбоксилирование калиевой соли 2,3,3,3-тетрафтор-2-иодпропионовой кислоты
В двугорлую колбу объёмом 100 мл, снабженную термометром и обратным холодильником, соединенным с холодильником глубокого охлаждения, охлаждаемого смесью сухой лед - ацетон, со склянкой Тищенко с конц. H2SO4, помещают 11 г (0.0355 моль) VII в 16 г ДМФА, раствор при перемешивании постепенно нагревают до 93С и перемешивают при этой температуре в течение 30 минут до окончания выделения газа. Реакционную массу охлаждают до комнатной температуры и выливают в двукратный объём 15% соляной кислоты, промывают 2x5 мл воды, сушат над CaCl2. Получают 3.2 г сырца, содержащего по данным ГХ: 26% IV, 74% V.
Благодарности
Работа выполнена при финансовой поддержке Министерства науки и высшего образования Российской Федерации. Строение полученных соединений изучено с использованием оборудования «Центра исследования строения молекул» ИНЭОС РАН (Москва, РФ).
Список литературы
- Tiers G., Journal of the American Chemical Society, 1960, 82(20), 5513.
- Патент США 3271441 (1966).
- V.C.R. McLoughlin, J.Thrower, Tetrahedron, 1969, 25(24), 5921-5940.
- Патент США 3351644 (1967).
- Патент США 3576885 (1971).
- Патент США 3862077 (1975).
- В.Э. Бойко, A. В. Едунов, С.М. Игумнов, Fluorine notes, 2016, 6(109), 5-6.
- Сборник «Синтезы фторорганических соединений», Часть 3, под ред. С.М. Игумнова, Э.В. Игумновой, Изд. «Тровант», М., 2015, 214с., 3.
- Патент Германии 3009760 (1981).
- Hauptschein M., Braid M., JACS, 1961, 83, 2383-2386.
- Petrov V.A., Krespan C.G., J. Org. Chem., 1996, 61, 9605-9607.
Статья рекомендована к публикации членом редколлегии к.х.н. М.А. Манаенковой
Fluorine Notes, 2022, 140, 7-8


