Поступило в редакцию: Февраль 2022
УДК 547.221
Fluorine Notes, 2022, 140, 1-2
Фторсодержащие простые эфиры
Сообщение 3. Получение присоединением спиртов к эпоксидам, взаимодействием фторолефинов и спиртов с формальдегидом, переэтерификацией, электрохимическим фторированием и рядом других методов.
С.В. Вершилова, В.В. Корнилов, А.С. Цырульниковаа,б, Л.М. Поповаа,б, Н.В. Лебедевa
а ФГУП «Научно-исследовательский институт синтетического каучука им. Академика С.В.Лебедева», ул. Гапсальская 1, Санкт-Петербург, 198035, Россия
б Санкт-Петербургский политехнический университет Петра Великого, ул. Новороссийская 48, Санкт-Петербург, 194021 Россия
Аннотация: В заключительной части обзора рассмотрены следующие методы получения фторсодержащих
простых эфиров: присоединение спиртов к эпоксидам, взаимодействие фторированных олефинов и спиртов
с формальдегидом, переэтерификация (transetherification) и электрохимическое фторирование.
Кроме
того, приведено краткое описание не вошедших в отдельные главы способов получения фторсодержащих
простых эфиров, а именно: свободно-радикальное присоединение фторсодержащих алкенов к простым эфирам,
фторирование элементным фтором и высшими фторидами металлов переменной валентности, реакции спиртов
с кетонами и карбоновыми кислотами, реакции перфторированных нитрозоалканов со спиртами и взаимодействие
спиртов c 2,4,6-трис-(2,2,2-трифторэтокси)-[1,3,5]триазином (TriTFET).
Ключевые слова: фторсодержащие простые эфиры, эпоксиды, формальдегид, переэтерификация, электрохимическое фторирование, трифторид кобальта, TriTFE.
Введение
В первых двух частях обзора [1] уже были рассмотрены наиболее известные методы синтеза фторсодержащих простых эфиров, а именно: реакция Вильямсона, присоединение алкенов и алкинов к спиртам, взаимодействие фторированных спиртов с диазометаном, присоединение полифторалкилиодидов к алкенам, межмолекулярная дегидратация, присоединение перфторалкилгипогалогенитов к алкенам и пиролиз производных перфтор-2-алкоксипропионовых кислот.
В третьей части обзора рассмотрено получение фторсодержащих простых эфиров присоединением фторсодержащих спиртов к эпоксидам, взаимодействием фторированных олефинов и спиртов с формальдегидом, переэтерификацией, а также методом электрохимического фторирования.
Кроме того, коротко рассмотрен ряд не вошедших в отдельные главы способов получения фторсодержащих простых эфиров, а именно: свободно-радикальное присоединение фторсодержащих алкенов к простым эфирам, фторирование элементным фтором и высшими фторидами металлов переменной валентности, реакции спиртов с кетонами и карбоновыми кислотами, реакции перфторированных нитрозоалканов со спиртами и взаимодействие спиртов c 2,4,6-трис-(2,2,2-трифторэтокси)-[1,3,5]триазином (TriTFET)
1. Присоединение спиртов к эпоксидам
Для синтеза частично фторированных простых эфиров могут использоваться как реакции фторсодержащих спиртов с углеводородными эпоксидами, так и реакции фторсодержащих эпоксидов со спиртами.
1.1. Взаимодействие фторсодержащих спиртов с эпоксидами
Подобно углеводородным аналогам взаимодействие фторсодержащих спиртов с эпоксидам в присутствии катализаторов кислого или основного характера приводит к образованию одной эфирной связи.
Полифторированные спирты общей формулы RF(CH2)nОН (n ≥ 1) взаимодействуют с этиленоксидом и пропиленоксидом с образованием фторсодержащих эфиров диолов.
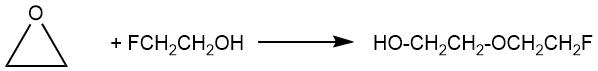
В 1949 г. работе И.Л.Кнунянца с сотр. [2] было сообщено о получении 2-фторэтилового эфира этиленгликоля (т.кип. 172-174°С) нагреванием смеси 2-фторэтанола с этиленоксидом при 170-180°С в течение 54 ч.
В 1957 г. М. Брей и П. Таррант описали присоединение полифторалканолов общей формулы RFCH2OH
(RF = CF3, C2F5, C3F7) к этиленоксиду
при температуре 70°C в присутствии гидроксида калия в автоклаве [3].
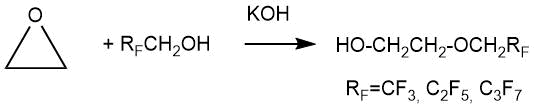
Методика присоединения полифторалканолов к этиленоксиду была следующая: охлажденный раствор 5 г КОН в 1,5 моль фторсодержащего спирта загружали в качающийся автоклав объемом 300 мл, который затем охлаждали смесью сухой лед – ацетон. Затем загружали 1 моль этиленоксида. Смесь выдерживали 4 ч при 70°С, после чего продукт выделяли дистилляцией. Характеристики полученных эфиров и их выходы приведены в Табл. 1.1.
Табл. 1.1.
|
Эфир |
Выход, % |
Т. кип., °С (мм.рт.ст.) |
nD25 |
d425,г/см3 |
|
CF3CH2OCH2CH2OH |
50 |
84(80) |
1,3502 |
1,2902 |
|
C2F5CH2OCH2CH2OH |
32 |
87 (84) |
1,3370 |
1,3806 |
|
C3F7CH2OCH2CH2OH |
62 |
91-92 (54) |
1,3300 |
1,4965 |
|
CF3(CH3)2COCH2CH2OH |
35 |
92 (77) |
1,3749 |
1,1931 |
|
(CF3CH2OCH2)2CHOH |
19 |
86 (16) |
1,3528 |
1,3890 |
|
(C3F7CH2OCH2)2CHOH |
24 |
112-115 (15) |
1,3338 |
1,5569 |
Методика, похожая на описанную М. Бреем и П. Таррантом, приведена в книге И.Л. Кнунянца и Г.Г. Якобсона
[4]. Снижение загрузки реагентов в 2 раза (для реактора объемом 250 мл), охлаждение жидким азотом
и вакуумирование реактора перед загрузкой этиленоксида, позволило повысить выход целевого 2-гидроксиэтил-1’,1’-дигидроперфторбутилового
эфира до 70%.

В 1957 году В. Гракаускас описал взаимодействие оксидов этилена и пропилена с 2-фтор-2,2-динитроэтанолом в водном гидроксиде натрия с получением соответствующих моноэфиров [5].
Так 2-фтор-2,2-динитроэтил 2-гидроксиэтиловый эфир был получен с выходом до 68% при добавлении 2-фтор-2,2-динитроэтанола и этиленоксида к водному раствору гидроксида натрия при 0÷5°C с последующей выдержкой реакционной смеси в течение 16 часов при 0°C.
2-Фтор-2,2-динитроэтил 2-гидроксипропиловый эфир был получен в аналогичных условиях с выходом около 25%. Выход целевого эфира увеличивается до 63% при увеличении мольного соотношения исходного 2-фтор-2,2-динитроэтанола к пропиленоксиду до 4:1.
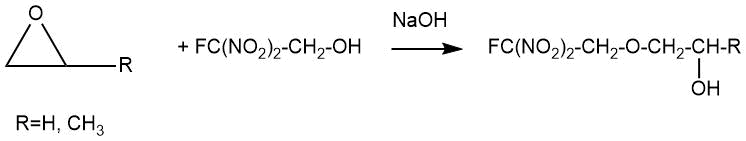
Проведение реакции 2-фтор-2,2-динитроэтанола с оксидом пропилена в условиях кислотного катализа (SnCl4) приводило к невысоким выходам эфира (12%).
В статье А.В. Фокина с сотр. [6] описано взаимодействие 2,2-дифтор-2-нитроэтанола (0,15 моль) с этиленоксидом
и пропиленоксидом (0,2-0,5 моль) в среде 5%-ного водного гидроксида натрия (0°С, 16 ч). В результате
реакций получали 2,2-дифтор-2-нитроэтил-2-гидроксиэтиловый и 2,2-дифтор-2-нитроэтил-2-гидроксипропиловый
эфиры с выходами 32 и 67% соответственно. В реакциях с этиленоксидом было отмечено образование побочного
2,2-дифтор-2-нитроэтилового эфира диэтиленгликоля (около 4%).
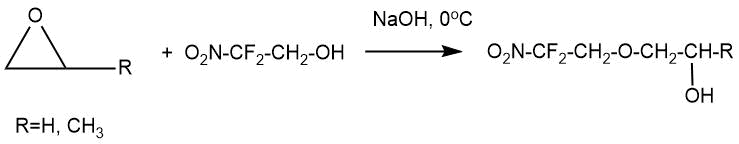
В патентах Бэнитта [7], [8], а также Бэнитта, Ченга и Джуса [9] показано, что моноаддукты могут быть получены взаимодействием фторсодержащих спиртов (CF3CH2OH, C3F7CH2OH) с этиленоксидом (мольное соотношение спирт : этиленоксид = 1 : 0,7-0,8) в присутствии каталитических количеств гидроксида натрия (в виде 50%-го водного раствора) в течение 5-7 ч при нагревании (70°С или кипячение). После дистилляции целевые эфиры (выход не указан) содержали около 10% исходных фторсодержащих спиртов.
Р. Томпсон и М. Хувер [10] получили 1,1-дигидроперфторгептиловый эфир этиленгликоля (т.кип. 212-213°С) нагреванием 1,1-дигидроперфторгептанола (2,99 моль) с этиленоксидом (1,63 моль) при 80°С. Замещением гидроксильной группы бромом (ZnBr2, Br2, H2SO4, 100-110°С) был получен 1-бромэтиловый эфир.
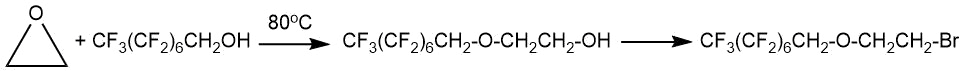
Взаимодействие фторалканолов с этиленоксидом или пропиленоксидом (ПО) может сопровождаться образованием олигомеров (полигликолей).
Так, образование фторалкилового эфира CF3(CF2)nCH2O(CH2CH2)mOH (n=7 среднее значение); m=10,6), согласно данным М. Хайека и Р. Моуди [11], происходило при нагревании фторсодержащего спирта с расчетным количеством этиленоксида в присутствии каталитического количества трифторида бора при 65-90°С в течение 10 ч.
Другие примеры приведены в патенте А Жура [12]. В присутствии эфирата трифторида бора при умеренной температуре
(40°С) происходит присоединение двух и более звеньев пропиленоксида к 2,2,3,4,4,4-гексафторбутанолу
CF3CFHCF2CH2OH при соотношении пропиленоксид : спирт = 3:1.
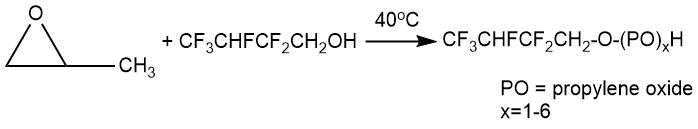
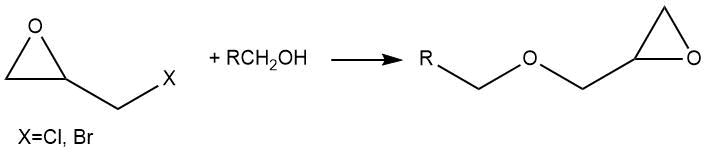
В реакциях фторсодержащих спиртов с 1-хлор-2,3-эпоксипропаном (эпихлоргидрином) и 1-бром-2,3-эпоксипропаном
(эпибромгидрином) конечными продуктами являются глицидиловые эфиры.
В работе М. Брея и П. Тарранта [3] приведено описание взаимодействия 1,1,1-трифторэтанола с 1-хлор-2,3-эпоксипропаном в присутствии гидроксида натрия как катализатора. При избытке гидроксида натрия конечным продуктом был глицидиловый эфир, который мог вступать в дальнейшую реакцию со спиртом с получением диэфира.
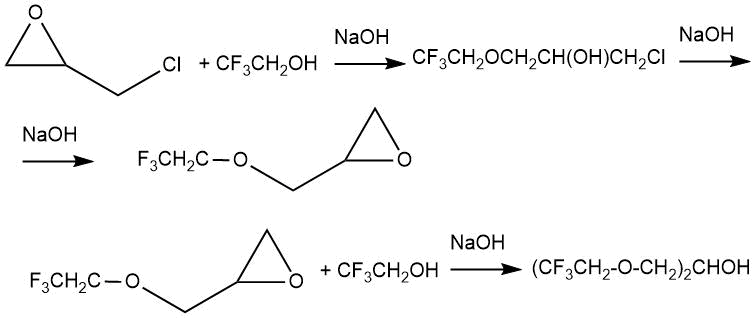
Так при проведении синтеза в охлажденном растворе гидроксида натрия в продуктах реакции было обнаружено 19÷31% глицидилового эфира и 19÷24% диэфира.
Введение в реакцию каталитических количеств пиридина позволяло получать в качестве простого продукта присоединения 2,2,2-трифторэтил-2-гидрокси-3-хлорпропиловый эфир с выходом 50%.
В. Гракаускас [5] получил 2-фтор-2,2-динитроэтилглицидиловый эфир (т.кип. 70-71°С при 0,1 мм.рт.ст., выход 31%) прибавлением эквимолярных количеств эпихлоргидрина и 2-фтор-2,2-динитроэтанола в избыток водного раствора гидроксида натрия (0-3°С, 48 ч). Действие 8% хлористоводородной кислоты (бромистоводородной, иодистоводородной и азотной) приводило к раскрытию оксиранового кольца [13] и образованию 1-хлор-2-(2-фтор-2,2-динитроэтокси)-2-пропанола (25°С, 16 ч выход 100%). Отмечено, что это вещество также является побочным продуктом при получении 2-фтор-2,2-динитроэтилглицидилового эфира из 2-фтор-2,2-динитроэтанола и эпихлоргидрина. При обработке раствора 1-хлор-2-(2-фтор-2,2-динитроэтокси)-2-пропанола в метаноле 85%-ным раствором гидроксида натрия в метаноле (22-25°С, по каплям, затем еще 15 мин) соответствующий глицидиловый эфир получался с выходом 95%.
1.2. Взаимодействие спиртов с фторсодержащими эпоксидами
Д. Сианези с соавторами в своей статье [14] и патенте [15] описали присоединение спиртов к оксиду гексафторпропилена (ОГФП, HFPO), которое приводило к образованию 2-алкоксипроизводных карбоновых кислот.
Так в результате взаимодействия метанола, этанола, 2,2,3,3-тетрафторпропанола и ряда других спиртов с ОГФП были получены сложные эфиры соответствующего спирта и 2-алкокситетрафторпропионовых кислот.
2ROH + HFPO → CF3CF(OR)COOR,
R = CH3, CH3CH2, ClCH2CH2 (CH3)2CH, HCF2CF2CH2, CH2=CHCH2
Если взаимодействие спиртов, которые не содержат в своем составе атомов фтора, с ОГФП легко протекает при комнатной температуре и атмосферном давлении, то реакция 2,2,3,3-тетрафторпропанола и ОГФП потребовала более жестких условий (автоклав, 80°C).
Также было приведено описание гидролиза синтезированных метил-2-метокситетрафторпропионата (мethyl 2-мethoxytetrafluoropropionate) и этил-2-этокситетрафторпропионата (ethyl 2-tthoxytetrafluoropropionatt) c получением соответствующих карбоновых кислот с высоким выходом.
CF3CF(OCH3)COOCH3 → CF3CF(OCH3)COOH (Выход 74%)
В статье Л. Брида, Р. Эллиотта и С. Кея [16] описано получение серии фторсодержащих сложных эфиров с простой эфирной связью в своей структуре присоединением спиртов общей формулы H(CF2)nCH2OH (n=4, 6) к ОГФП при 80°С в присутствии гидроксида натрия.
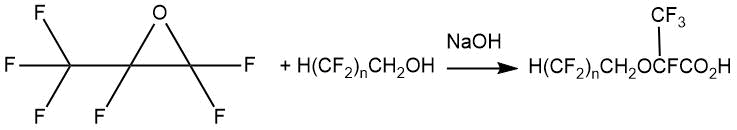
Выход таких кислот с простым эфирным фрагментом в своей структуре в случае 1,1,2-тригидроперфторпентанола составлял 46%, в случае 1,1,2-тригидроперфторгептанола – 67%.
В дальнейшем восстановлением 2-алкокси-2,3,3,3-тетрафторпропионовых кислот боргидридом натрия в тетрагидрофуране были получены соответствующие 2-алкокси-2,3,3,3-тетрафторпропанолы (для n=4 выход 32%; n=6 выход 39%).
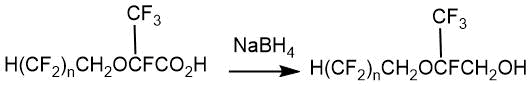
2. Образование фторсодержащих простых эфиров в реакциях с формальдегидом
2.1. Взаимодействие фторсодержащих олефинов с формальдегидом
Взаимодействие фторсодержащих олефинов с формальдегидом в зависимости от условий реакции может приводить к различным продуктам.
В 1949 г. Д.Кофман с соавт. [17] показал, что продуктом реакции параформальдегида и тетрафторэтилена (ТФЭ) в среде концентрированной серной кислоты (80°С, 15 ч) является 2,2,-дифтор-3-гидроксипропионовая кислота с выходом около 25%.
CF2=CF2 + CH2O + H2O → [HOCH2CF2CF2OH] → HOCH2CF2COOН
В 1952 г. эта же реакция была изучена МакБи с сотр. [18]. Авторами была получена дифторгидрокспропионовая кислота с выходом 20% (среда 95% серная кислота, 85°С).
В то же время в среде безводного фтористого (или хлористого) водорода реакции взаимодействия фторсодержащих олефинов с формальдегидом могут приводить как к получению простых эфиров различного строения, так и спиртов.
В 1963 г. в статье В. Вейнмайра [19] было приведено описание взаимодействия формальдегида с фторсодержащими олефинами в среде фтористого водорода.
На основании данных ЯМР-спектроскопии В. Вейнмайр установил, что растворение параформальдегида в безводном фтористом водороде сопровождается выделением воды и образованием соединений, содержащих FCH2-группы. В. Вейнмайром было сделано предположение, что гипотетическими интермедиатами, вступающими в дальнейшие взаимодействия с алкенами, являются фторэтанол и дифторметиловый эфир.
СН2О + HF → FH2COH
2СН2О + 2HF → FH2COCH2F
В процессе исследований было обнаружено, что продуктами взаимодействия формальдегида с фторэтиленом и 1,1-дифторэтиленом при низких температурах являлись симметричные простые эфиры. В случае тетрафторэтилена в качестве основных продуктов реакции получались 2,2,3,3,3-пентафторпропан-1-ол и фторметил-2,2,3,3,3-пентафторпропиловый эфир. Причем при низких температурах реакции (~20°C) в качестве основного продукта реакции получался фторметил-2,2,3,3,3-пентафторпропиловый эфир. При более высоких температурах синтеза (50÷100°C) основным продуктом реакции был 2,2,3,3,3-пентафторпропан-1-ол.
Реакция с гексафторпропиленом требовала более высоких температур (выше 100°C) и приводила к образованию исключительно 2,3,3,3-тетрафтор-2-трифторметилпропанола. Результаты исследований Вейнмайра приведены в Табл. 2.1.
Табл. 2.1. Результаты исследований взаимодействия параформальдегида с фторсодержащими алкенами в среде фтористого водорода.
|
Aлкен |
Алкен/Н2С=О/HF |
T, °С; τ, ч |
Продукты |
|
FCH=CH2 |
160/30/200 |
минус 40 ÷ 100°С; |
(HCF2CH2CH2)2O |
|
F2C=CH2 |
271/120/560 4,2/4,0/изб. |
0÷ 10°С; |
(CF3CH2CH2)2O |
|
F2C=CF2 |
Изб./6/изб. |
50°С, 24 ч |
CF3CF2CH2OCH2F CF3CF2CH2OH (CF3CF2CH2O)2CH2 (CF3CF2CH2OCH2)2O |
|
20°С, 24 ч |
CF3CF2CH2OCH2F CF3CF2CH2OH |
||
|
CF3CF=CF2 |
160/30/200 |
160°С, 8 ч |
CF3CF(CF3)CH2OH |
Результаты работы В. Вейнмайра, были подтверждены Л.С. Германом и И.Л. Кнунянцем, которые в аналогичных условиях из 1,1-дифторэтилена и формальдегида получили 3,3,3-трифторпропиловый эфир с выходом 47% [20].
СН2=CF2 + CH2O + HF → О(CH2CH2CF3)2
В другой своей работе Л.С. Герман и И.Л. Кнунянц [21] предложили возможный механизм реакции, который заключается в том, что под действием безводного HF происходит ионная диссоциация молекулы промежуточного фторметанола (FH2COH) по связи C-F с образованием оксиметилкатиона. На следующей стадии оксиметилкатион осуществляет электрофильную атаку на кратную связь олефина (Схема 2.1).
В случае 1,1-дифторэтилена реакция не останавливается на стадии получения фторметил-3,3,3-трифтопропилового эфира, а происходит присоединение второй молекулы олефина с образованием ди-3,3,3-трифторпропилового эфира. Авторы статьи объяснили это тем, что в фторметил-3,3,3-трифтопропиловом эфире трифторметильная группа находится на достаточно большом удалении от атома кислорода, её индукционное влияние слабо, поэтому легко происходит диссоциация по связи CF (Схема 2.2).
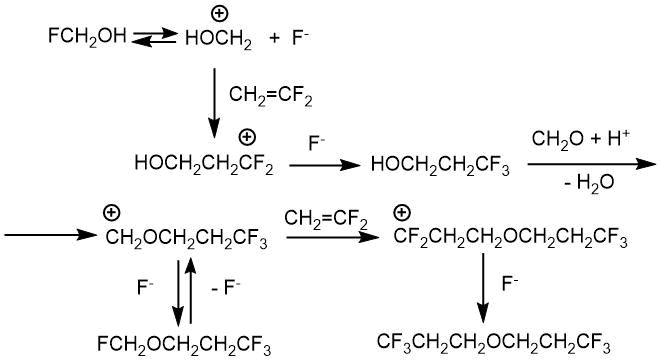
Схема 2.1.
Отличие взаимодействия формальдегида с тетрафторэтиленом, в котором основными продуктами реакции являются фторметил 3,3,3,2,2-пентафторпропиловый эфир и 2,2,3,3,3-пентафторпропанол, состоит в том, что сильный индукционный эффект пентафторпропильной группы препятствует диссоциации связи CF во фторметильной группе. По этой причине присоединение второй молекулы олефина к фторметил-3,3,3,2,2-пентафторпропиловому эфиру не происходит (Схема 2.2).
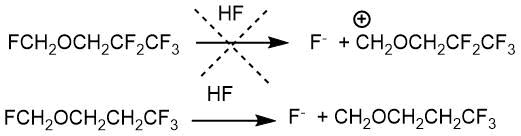
Схема 2.2
Кроме того, авторы привели описание взаимодействия параформальдегида с трифторэтиленом при 0÷-5°C. В этом случае в продуктах реакции присутствовали как симметричный ди-2,3,3,3-тетрафторпропиловый эфир c выходом 20%, так и 2,3,3,3,-тетрафторпропанол (выход 32%).
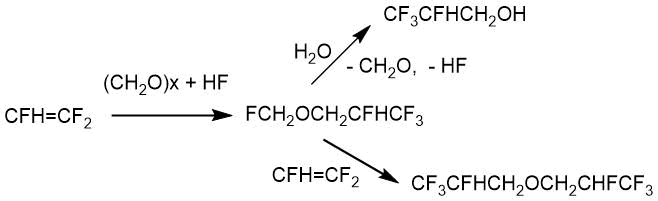
В патенте В. Вейнмайра [22] были представлены зависимости состава продуктов реакции ТФЭ с параформальдегидом от содержания воды во фтористом водороде. В среде безводного фтористого водорода основными продуктами были трифторметил-2,2,3,3,3-пентафторпропиловый эфир и 2,2,3,3,3-пентафторпропан-1-ол. С увеличением содержания воды во фтористом водороде содержание эфира снижалось. Результаты приведены в Табл. 2.2.
Табл. 2.2. Соотношение продуктов реакции параформальдегида с ТФЭ в зависимости от содержания воды во фтористом водороде [19].
|
Состав HF |
Температура, °С |
Давление, МПа |
Соотношение CF3CF2CH2OCH2F : CF3CF2CH2OH |
|
Безводный HF |
50 |
2,07 |
181:125 |
|
Безводный HF |
75 |
2,07 |
104:156 |
|
90% HF, 10% H2O |
50 |
2,76 |
28:222 |
|
80% HF, 20% H2O |
75 |
2,76 |
Только CF3CF2CH2OH |
В патенте Х. Яйле [23] описано взаимодействие тетрафторэтилена с формальдегидом в присутствии фторида цезия (триэтиленгликоль, 100°С, 5 ч), которое приводит к образованию с высоким выходом 2,2,3,3,3-пентафторпропанола.
CF2=CF2 + CH2O → CF3CF2СН2OH
2.2. Взаимодействие фторсодержащих спиртов с формальдегидом
Взаимодействие фторсодержащих спиртов с формальдегидом в присутствии кислотных катализаторов (HCl, HF, H2SO4) может приводить к получению простых эфиров.
В статье И.Л. Кнунянца [24] рассматривалось взаимодействие 2-фторэтанола с триоксаном (триоксиметиленом, тримером формальдегида). Было показано, что в зависимости от соотношения реагентов и условий проведения реакции образуются разные продукты.
Так при пропускании сухого HCl в смесь 0,3 моля 2-фторэтанола и 0,1 моля триоксана до полного растворения второго реагента (при охлаждении льдом и дальнейшем выдерживании в течение 24 часов при комнатной температуре) был получен хлорметил-2-фторэтиловый эфир с выходом 60%.
FCH2CH2OH + (CH2O)3 → FCH2CH2OCH2Cl
В то же время при взаимодействии 0,47 моля 2-фторэтанола и 0,05 моля триоксана (продувка сухим HCl до полного растворения второго реагента при охлаждении льдом и выдерживание 72 ч при комнатной температуре) был получен бис-(2-фторэтокси)метан с выходом 50% (т.кип. 162-164°С):
FCH2CH2OH + (CH2O)3 → FCH2CH2OCH2OCH2CH2F
Г. Олах с сотр. [25] также изучали взаимодействие триоксана с 2-фторэтанолом. Суспензию триоксана (4,5 г) в 2-фторэтаноле (30 г) продували безводным хлористым водородом с получением бис-(2-фторэтокси)метана (9,5 г). В другом примере авторы использовали тример ацетальдегида (Paraldehyde) и получили в качестве продукта реакции 1,1-бис-(2-фторэтокси)этан ((FCH2CH2O)2CHСН3).
Примеры синтеза ряда хлорметиловых эфиров фторсодержащих спиртов в подобных условиях приведены в патенте Г. Тесоро и Р. Ринга [26]. Смесь спирта и параформальдегида в среде диметоксиэтана и/или петролейного эфира обрабатывали сухим хлористым водородом при перемешивании. Затем смесь выдерживали в течение 5-10 ч. при умеренной температуре (5-20°С). Результаты представлены в Табл. 2.3.
Табл. 2.3.
|
Спирт |
Соотношение (моль) Спирт/(CH2O)n |
T, °С; τ, ч |
Основные продукты, выход (%) |
|
CF3CH2OH |
1,05/4,3 |
5 ÷ 10°С |
CF3CH2OCH2Cl (50%) |
|
CF2H(CF2)5CH2OH |
1,4/7,7 |
5°С |
CF2H(CF2)5CH2OCH2Cl (40%) |
|
CF3(CF2)6CH2OH |
0,25/1,37 |
0 ÷ 10°С |
CF3(CF2)6CH2OCH2Cl (47%) |
|
CF2H(CF2)9CH2OH |
0,86/4,7 |
5 ÷ 15°С |
CF2H(CF2)7CH2OCH2Cl (97%) |
|
OHCH2(CF2)3CH2OH |
0,3/3 |
5 ÷ 10°С |
CH2ClOCH2(CF2)3CH2OCH2Cl |
В.А. Комаров с соавт. [27] показали, что взаимодействие 2,2-дифтор-2-нитроэтанола с формальдегидом и хлористым водородом (ZnCl2, минус 10°С, 2 ч) приводит к получению 2,2-дифтор-2-нитроэтилхлорметилового эфира (т. кип. 54°С/15 мм Hg) с выходом 54,5%.
O2NCF2CH2OH + CH2O + HCl → O2NCF2CH2OCH2Cl
При более высокой температуре (от комнатной до 60°С) продуктом реакции этих же реагентов является гидроксиметил-2,2-дифтор-2-нитроэтиловый эфир (т.кип. 116°С/3 мм Hg) с выходом 76,5% [28].
O2NCF2CH2OH + CH2O + HCl → O2NCF2CH2OCH2ОН
В статье Х.Адольфа и М.Камлета [29] описана реакция 2-фтор-2,2-динитроэтанола с полиформальдегидом в кислой среде. При использовании концентрированной серной кислоты и температуре реакции 0-10°С получается бис-(2-фтор-2,2-динитроэтокси)метан с выходом 80%.
(O2N)2CFСН2ОН + (СН2О)n → (O2N)2CFСН2ОCH2OCH2C(NO2)2F
При использовании избытка формальдегида и более низкой концентрации серной кислоты (80-90%) наблюдается образование гомологического ряда продуктов (O2N)2CFСН2О(CH2O)nCH2C(NO2)2F (n=2-4).
Взаимодействие формальдегида с 2,2,2-трифторэтанолом и 2,2,3,3-тетрафторпропанолом в среде серной кислоты описано в патенте М. Хилл и К. Шипп [30]. Авторами цитируемой работы была определена зависимость выхода эфира от концентрации серной кислоты (от 80% H2SO4 до 10% олеума). Было установлено, что максимальный выход эфира достигается при использовании 96% H2SO4. Синтезы проводили в течение 1 часа при комнатной температуре в условиях перемешивания. В результате были получены бис-(2,2,2-трифторэтокси)метан с выходом 68% и бис-(2,2,3,3-тетрафторпропокси)метан с выходом 63,4%.
А.Я. Запеваловым с соавт. [31] рассмотрен метод получения бис-2,2,3,3,4,4,5,5-октафторпентилокси)метана с выходом 73-91% нагреванием смеси 2,2,3,3,4,4,5,5-октафторпентанола и формальдегида (мольное отношение 2:1) в о-ксилоле (п-ксилоле) в присутствии толуолсульфокислоты.
CF2H-CF2CF2CF2CH2-OH + CH2O → CF2H-CF2CF2CF2CH2-O-CH2-O-CH2CF2CF2CF2CF2H
Получение 2-фтор-3-гидрокси-н-пропилметилового эфира через промежуточный синтез бис-(2-фтор-3-хлорпропокси)метана описано в статье Л.С.Богуславской с соавт. [32]. Бис-(2-фтор-3-хлорпропокси)метан образуется при взаимодействии 2-фтор-3-хлор-1-пропанола с параформальдегидом в присутствии п-толуолсульфокислоты в бензоле с выходом 76%.
CH2ClCHFCH2OH + (CH2O)n → (CH2ClCHFCH2O)2CH2 → (CH3OCH2CHFCH2O)2CH2 → CH3OCH2CHFCH2OH
М. Хилл и Л. Росс сообщали [33] о получении бис-(2,2-дифтор-2-нитроэтилокси-)метана с выходом 60% при нагревании 2,2-дифтор-2-нитроэтанола (0,07 моль) с параформальдегидом (0,016 моль) в среде 90%-ной H2SO4.
O2NCF2CH2OH + HCHO → O2NCF2CH2OCH2OCH2CF2NO2
Полифторалкилхлорметиловые эфиры были получены А.Альбрехтом [34] при обработке сухим хлористым водородом смеси полифторированного спирта с небольшим избытком параформальдегида в бензоле с последующей отгонкой летучих компонентов.
C8F17(CH2)nOH +(CH2O)m → C8F17(CH2)nOCH2Cl, n= 5, 11.
3. Переэтерификация
В патенте Коддинга [35] описан синтез гептафторбутилвинилового эфира с выходом 32% взаимодействием гептафторбутанола с винилацетатом в присутствии ацетата ртути в среде серной кислоты.
Другой пример синтеза простого винилового эфира реакцией 2,2-динитро-2-фторэтанола с винилацетатом в присутствии ацетата ртути и серной кислоты (0°С, 16 ч, 51%) приведен в патенте Адольфа [36].
(O2N)2CFCH2OH + СН2=СНОС(О)СН3 → (O2N)2CFCH2OСН=СН2
В патенте Л.Крокса [37] приведен пример реакции 2,2,2-трифторэтанола с винилацетатом с получением 1,1-бис-(2,2,2-трифторэтокси)этана (HgO, BF3·Et2O, 30-50°С, 2 ч, выход 61,9%). Последующий пиролиз 1,1-бис-(2,2,2-трифторэтокси)этана (катализатор AgNO3, Fe2O3, Pt-асбест, 340-390°С) приводит к получению 2,2,2-трифторэтилвинилового эфира с выходом около 88%.
СF3CH2OH + СН2=СНОС(О)СН3 → СH3CH(OCH2CF3)2 → СF3CH2OСН=СН2
В работе [38] были рассмотрены реакции фторсодержащих спиртов общей формулы H(CF2)nCH2OH (n=4,6,8) с винилацетатом в присутствии солей Hg (II) в качестве катализатора с получением смеси продуктов: винилового эфира, его ацетилпроизводного и ацеталя.
H(CF2)nCH2OH + СН2=СНОС(О)СН3 → H(CF2)nCH2OCH=CH2 +
+ CH3CH(-OC(O)CH3)OCH2(CF2)nH + CH3CH(OCH2(CF2)nH)2
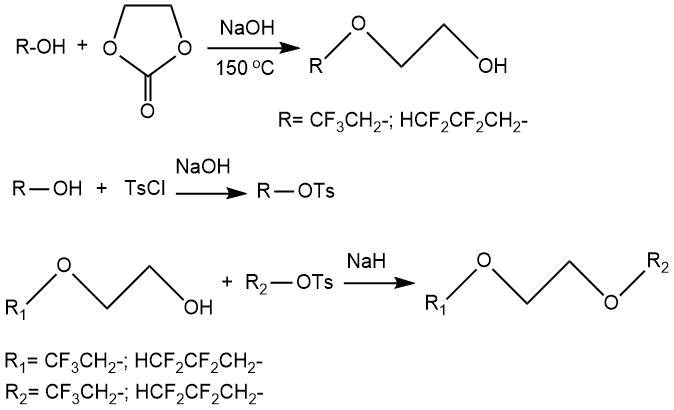
В статье Б. Мандала [39] и патенте В. Тортелли [40] описано получение 2,2,2-трифторэтил 2-гидроксиэтилового эфира взаимодействием этиленкарбоната с 2,2,2-трифторэтанолом в присутствии гидроксида натрия (тетраглим, 150°С, 4 ч, выход 76%).
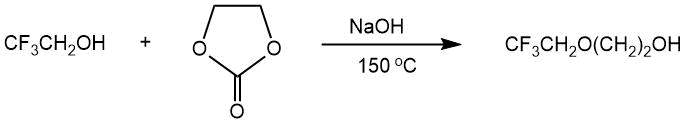
В работе [41] рассмотрено получение моноэфиров этиленгликоля CF3CH2O(CH2)2OH
и CНF2CF2CH2O(CH2)2OH через реакции соответствующих
спиртов с этиленкарбонатом. Дальнейшая обработка полученных моноэфиров тозилатами исходных фторсодержащих
спиртов приводила к образованию 1,2-бис-(2,2,2-трифторэтокси)этана (CF3CH2O(CH2)2OCH2CF3)
и 1,2-бис-(2,2,3,3-тетрафторпропокси)этана (CНF2CF2CH2O(CH2)2OCH2CF2CHF2)
с выходами 28-42%.
Б. Ботевин и Б. Юссеф исследовали реакции 1,1,2,2,-тетра-H-перфтороктан-1-ола с этилвиниловым эфиром в присутствии ацетата ртути (или комплекса Pd (II) [diacetato-(1,10-phenanthroline) palladium (II)]), которые приводят к образованию 1,1,2,2,-тетра-H-перфтороктил винилового эфира [42].
C6F13C2H4OH + C2H5OCH=CH2 → C6F13C2H4OCH=CH2.
Х.Адольфом и М.Камлетом было показано, что взаимодействие 2-фтор-2,2-динитроэтанола с 5-кратным избытком диметоксиметана в присутствии эфирата трифторида бора (кипячение, 3 ч) ведет к образованию 1,1-динитро-1-фтор-2-(метоксиметокси)этана [29].
CF(NO2)2CH2OH + CH3OCH2OCH3 → CF(NO2)2CH2OCH2OCH3
В среде концентрированной серной кислоты (т. комн., 15 ч,) реакция уже приводила к получению бис(2-фтор-2,2-динитроэтокси)метана с выходом 64%.
CF(NO2)2CH2OH + CH3OCH2OCH3 → CF(NO2)2CH2OCH2OCH2C(NO2)2F
Дж. Бинчем с соавт. [43] при изучении конденсации 2,2-дифтор-1,3-пропандиола с диметоксиметаном (нагревание в присутствии полистиролсульфокислоты, Amberlyst-15) было отмечено, что помимо целевого 5,5-дифтор-1,3-диоксана, среди продуктов присутствуют линейные соединения с эфирной группой СH3OCH2OCH2CF2CH2OH и СH3OCH2OCH2CF2CH2OCH2OCH3.
В статье К. Петрова с соавт. [44] приведено описание взаимодействия ряда фторсодержащих спиртов с ортоэфирами с получением простых эфиров.
RFOH + RC(OR’)3 → RFOR’,
Где RF= CF3CH(OH), (CF3)2C(OH), (ClCF2)2C(OH); R= H, CH3; R’=CH3, C2H5.
4. Получение фторсодержащих простых эфиров методом электрохимического фторирования
Полностью фторированные простые эфиры образуются с умеренным выходом при электрохимическом фторировании (ЭХФ, ECF) соответствующих углеводородных аналогов:
CnH2n+1OCmH2m+1. → CnF2n+1OCmF2m+1
В качестве побочных продуктов получаются соединения, которые образуются вследствие разрыва связей CO и CC, причем с увеличением молекулярного веса исходного диалкилового эфира содержание побочных продуктов увеличивается.
Метод ЭХФ был разработан Дж. Саймонсом в конце 40-х годов 20 столетия [45]. Первое упоминание о получении фторсодержащих простых эфиров этим методом приведено в патенте самого Дж. Саймонса в 1950 г [46]. Было приведено описание получения перфтордиметилового, перфтордиэтилового, перфтор-н-дибутилового, перфтор-н-диамилового, перфтор-н-дигексилового,, и ряда других эфиров из исходных углеводородных прекурсоров, а также перфтор-1,2-бис-(метокси)этана из 1,4-диоксана.
Методика получения была следующей: исходный углеводородный эфир растворяли в безводном фтористом водороде и осуществляли процесс ЭХФ в электрохимической ячейке (cell) с никелевым анодом и железным катодом при атмосферном давлении, температуре 0°C, напряжении 4÷6 вольт и силе тока около 20 A/ft2. По мере срабатывания исходного углеводородного эфира в ходе процесса в ячейку добавляли новые порции сырья. Выход целевых продуктов был не указан.
В патенте З. Беннингера (S. Benninger) [47] описан процесс получения перфторированных эфиров многоатомных спиртов.
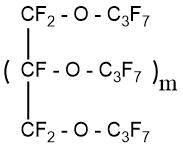
m=1-4
Процесс состоял из двух стадий:
- На первой стадии проводили синтез частично фторированных эфиров из многоатомного спирта и гексафторпропилена
- полученный частично фторированный эфир подвергали ЭХФ в электролизере Саймонса с получением перфторированных соединений.
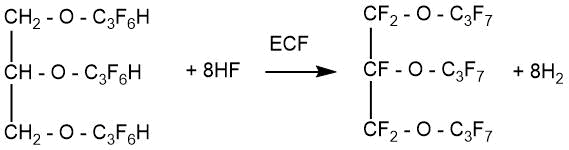
О получении вместе с перфторкарбоновыми кислотами перфторированных эфиров с невысокими выходами при ЭХФ диэтилового, дипропилового и ряда других простых эфиров сообщалось в статье японских исследователей [48].
В работе К. Оказаки с сотр. [49] сообщалось о ЭХФ в среде фтористого водорода хлорметил метилового эфира, трихлорметил метилового эфира, 2,2,2,-трихлорэтил метилового эфира и ряда других хлорсодержащих простых эфиров. Было установлено, что если атом хлора находился у α-атома углерода, то он замещался на фтор при ЭХФ. Если же атом хлора находился в β-положении, то он оставался неизменным с получением β-хлорированных полифторсодержащих простых эфиров. При каждом синтезе получалась сложная смесь продуктов с преобладанием продуктов деструкции.
В патенте Д. Хансена (J. C. Hansen) [50] описано ЭХФ метил 1,1,3,3,3-пентафтор-2-(трифторметил)пропилового эфира ((CF3)2CHCF2OCH3) и этил 1,1,3,3,3-пентафтор-2-(трифторметил)пропилового эфира ((CF3)2CHCF2OC2H5). Оба этих эфира являются продуктами утилизации токсичного перфторизобутилена (PFIB), который образуется как побочный продукт при синтезе гексафторпропилена.
В таблице 4.1 приведен состав продуктов после ЭХФ метил 1,1,3,3,3-пентафтор-2-(трифторметил)пропилового эфира. Соотношение полностью фторированного эфира и эфира с остающимся атомом водорода в положении 2 пропильного фрагмента можно было изменять от 10:1 до 1:1 варьированием концентрации фтористого водорода в электропроводящем растворе.
Таблица 4.1.
|
Компонент |
Т. кип. °C |
Выход, % от теорет. |
|
(CF3)2CFCF2OCF3 |
33 |
35÷40 |
|
(CF3)2CHCF2OCF3 |
46 |
25÷30 |
|
(CF3)2CFCOF |
0 |
5÷10 |
|
(CF3)2CHCOF |
15 |
5 |
|
(CF3)2CFCF3 |
0 |
5÷10 |
|
(CF3)3CH |
12 |
10÷15 |
|
(CF3)2CHCF2OCHF2 + (CF3)2CHCF2OCH2F |
- |
<5 |
|
(CF3)2CHCF2OCH3 |
69,5-70 |
5 |
Для улучшения параметров процесса ЭХФ применяют специальные «активные добавки» к электролиту, которые позволяют стабилизировать сам процесс ЭХФ и проводить его в непрерывном режиме.
В статье Г.И. Кауровой [51] приведены результаты сравнения эффективности добавок на процесс синтеза дибутилового эфира (Таблица 4.2).
Таблица 4.2. ЭХФ дибутилового эфира (C4H9)2O.
|
Добавка |
i, А/см2 |
U, В |
Q, А*ч/л |
Выход по току сырца, % |
Коррозия анодов, г/А*ч |
|
н-бутилмеркаптан 15% |
0.02 |
4.6-5.0 |
4000 |
64 |
<0.001 |
|
гидрохинон - 3.5 % |
0.02 |
4.6-5.5 |
1600 |
33 |
0.03 |
|
иод (I2) - 4 % |
0.01 |
4.9-7.0 |
<1000 |
25 |
0.03 |
Как видно из результатов таблицы наилучшие результаты были получены с применением н-бутилмеркаптана.
В работах В.А. Маталина [52] [53] исследовано влияние третичных аминов, использованных в качестве добавок в электролит в процессе электрохимического фторирования различных органических соединений. Были определены оптимальные концентрации фторируемого соединения и амина в электролите (5-15 % масс). Сравнительные результаты ЭХФ с применением н-бутилмеркаптана и триаллиламина ((C5H5)3N) приведены в таблице 4.3.
Таблица 4.3. Электрохимическое фторирование ((C4H9)2O (I – 0,03 А/см2).
|
Исходные соединения |
Выход по току, % |
Основные продукты фторирования, % масс. |
|
|
(С4Н9)2О |
C5F10NF2 + C5F12 |
||
|
(С4Н9)2О + C4H9SH |
49 |
- |
- |
|
(С4Н9)2О +(C5H5)3N |
46 |
50 |
40 |
При ЭХФ фторангидридов карбоновых кислот линейного строения наряду с получением перфторированных производных карбоновых кислот наблюдается образование циклических перфторированных эфиров (смесь перфтор-2-алкилоксоланов и перфтор-2-алкилоксанов.
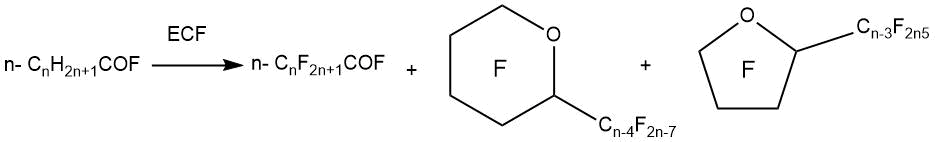
Например, в работе Г. Гамбаретто с сотрудниками [54] показано, что при ЭХФ фторангидрида октановой кислоты (октаноилфторида) образуются продукты его циклизации с высокими выходами (до 60%). Было обнаружено, что происходит образование как перфторпроизводных α-замещенных оксанов (до 69% в смеси), так и в меньшей степени перфторпроизводных α-замещенных оксаланов (до 23% в смеси). Образование циклических соединений в процессе ЭХФ можно было снизить за счет использования в качестве сырья частично фторированных октаноилфторидов.
С другой стороны, при ЭХФ производных циклических α-замещенных тетрагидропирана (оксана) также не удается избежать процессов изомеризации. В работе Т.Абэ с сотрудниками [55] показано, что в ходе ЭХФ α-замещенных тетрагидропиранов идут как процессы перегруппировок с образованием пятичленных циклов, так и раскрытия оксанового кольца с образованием линейных структур.
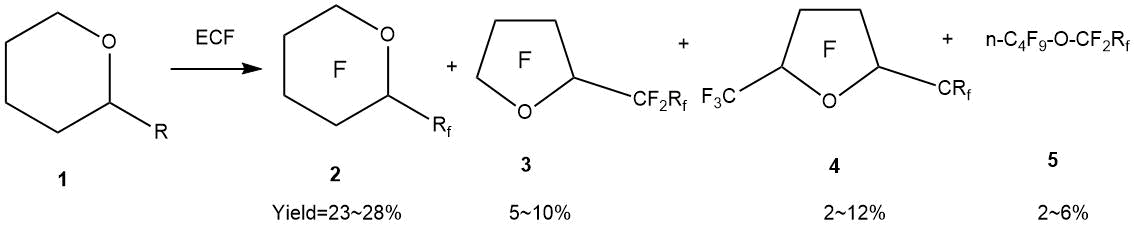
a: R=C2H5, Rf=C2F5; b: R=n-C3H7, Rf =n-C3F7; c: R=iso- C3H7, Rf =iso- C3F7;
d: R=n-C4H9, Rf =n-C4F9; e: R=n-C5H11, Rf =n-C5F11;
В статье Т. Фучигами с сотр. [56] описано ЭХФ некоторых эфиров и краун-эфиров при использовании в качестве электролита комплексов триэтиламина с фтористым водородом. При ЭХФ диметоксиэтана в Et3N·5HF или Et4NF·4HF наблюдалось образование смеси двух монофторированных продуктов.
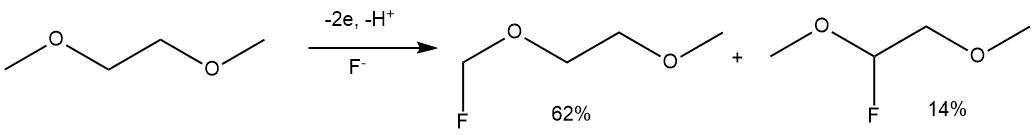
ЭХФ в схожих условиях диметилового эфира диэтиленгликоля (диглима) приводило к получению исключительно монофторпроизводного у терминального атома углерода (выход 55%, электролит Et3N·5HF).
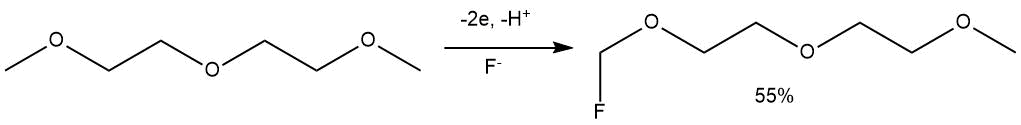
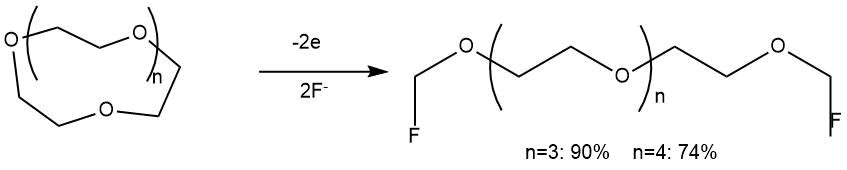
В то же время ЭХФ краун-эфиров приводило к преимущественному разрыву связи С-С с образованием α,ω-дифторзамещенных
продуктов.
5. Прочие реакции получения простых фторсодержащих эфиров
5.1. Свободно-радикальное присоединение фторсодержащих алкенов к простым эфирам
Реакции свободно-радикального присоединения фторсодержащих алкенов с простыми эфирами проводят в условиях γ-облучения или в присутствии пероксидов.
В статье Х. Мураматсу с сотр. [57] была описана реакция присоединения 1,1,2-трихлорфторэтилена к диэтиловому эфиру. Исходную смесь алкена и эфира помещали в стеклянную трубку и подвергали γ-облучению при комнатной температуре в течение 312 часов. В результате была получена смесь 1-метил-2-фтор-2,3,3-трихлорпропил этилового эфира с выходом 16% и бис-1-метил-2-фтор-2,3,3-трихлорпропилового эфира с выходом 26%.
В другой статье этих же авторов [58] изучено присоединение полифторалкил этиловых эфиров к гексафторпропену. Реакцию проводили в автоклаве в условиях γ-облучения. Так в результате реакции (rt, 1030 час.) 2,2-дифторэтил этилового эфира (37 г) и гексафторпропена (137 г) был получен 2,2,-дифторэтил 1-метил-2,2,3,4,4,4-гексаторбутиловый эфир с выходом 64%.
В работе Р. Чамберса с соавт. [59] исследованы реакции присоединения ряда ациклических простых эфиров к гексафторпропену в одинаковых условиях (избыток эфира, γ-облучение, 18°C).
В зависимости от природы заместителя в эфире в продуктах реакции наблюдали продукты присоединения одной, двух или даже трех молекул гексафторпропена (Табл. 5.1.).
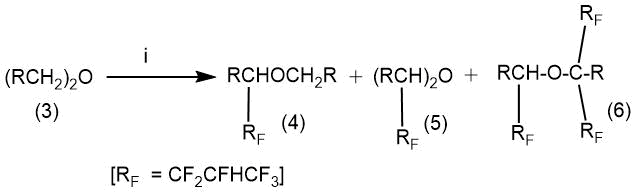
Таблица 5.1.
|
Продукты (% состав) |
Конверсия % |
|||
|
(3a) R=H |
(4a) (100) |
- |
- |
70 |
|
(3b) R=Me |
(4b)(47) |
(5b)(53) |
- |
100 |
|
(3c) R=Et |
(4c) (30 |
(5c) (70) |
- |
80 |
|
(3d) R= n-Pr |
(4d) (23 |
(5d) (40) |
(6d)(40) |
70 |
5.2. Фторирование элементным фтором и высшими фторидами металлов переменной валентности
Метод прямого фторирования элементным фтором имеет очень ограниченное применение для получения фторсодержащих соединений, в том числе и фторированных эфиров, вследствие высокой реакционной способности элементного фтора. В большинстве случаев выход целевых соединений низок, а в качестве основных продуктов реакции получаются продукты деструкции.
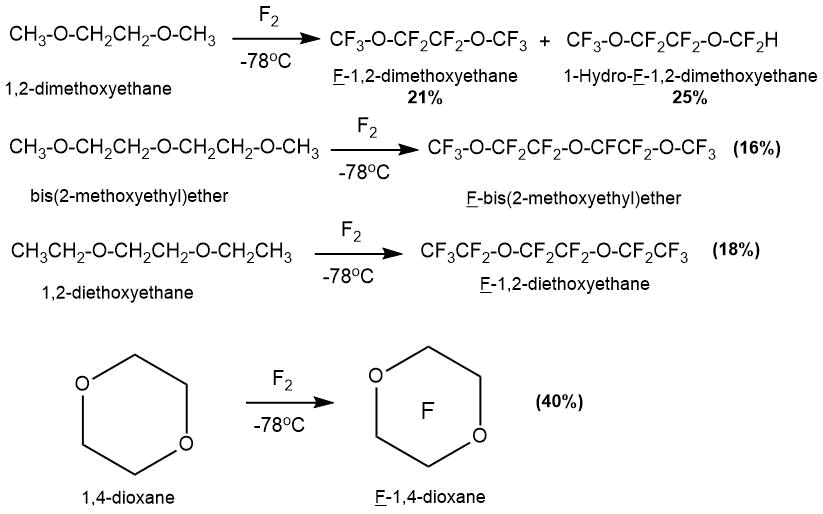
В 1975 г. в статье Р. Лагоу с сотр. [60] было описано получение ряда перфторированных соединений с простой эфирной связью из углеводородных прекурсоров (1,2-Диметоксиэтана, ди-2-метоксиэтилового эфира, 1,2-диэтоксиэтана).
Синтез осуществляли в мультизонном криогенном реакторе с градиентом температур по длине реактора. Температура первой зоны составляла минус 78°C, температуры последующих зон варьировались для разных продуктов. Следует отметить, что несмотря на применение специально разработанной конструкции реактора и низких температур процесса, выход целевых продуктов был невысок.
Большинство попыток получения фторированных простых эфиров с использованием высших фторидов металлов переменной валентности в качестве фторирующих агентов заканчивались неудачно [61]. В процессе фторирования эфирная группа, впрочем, как и другие функциональные группы, подвергалась деструкции.
В 1974 г. в работе Дж. К. Татлоу с соавт. [62] были приведены результаты фторирования диэтилового и метил этилового эфиров с применением трифторида кобальта (CoF3) при 60÷80°C и тетрафторкобальтата калия (potassium tetrafluorocobaltate, KCoF4) при 200°C. В обоих случаях были получены продукты деструкции вместе со сложной смесью полифторированных эфиров. Например, в реакции трифторида кобальта с диэтиловым эфиром были идентифицированы 1,2-дифторэтан, 1,1,2,2-тетрафторэтил 1,2,2-трифторэтиловый эфир, бис-1,2,2-трифторэтиловый эфир, 1,2-дифторэтил 1,2,2-трифторэтиловый эфир.
В работе Р. Чамберса и Б. Гривсона [63] описано получение перфторированных простых эфиров исчерпывающим фторированием продуктов свободно-радикального присоединения простых эфиров (см. 5.1.) к фторсодержащим алкенам. В качестве побочных продуктов образовывались перфторалканы.
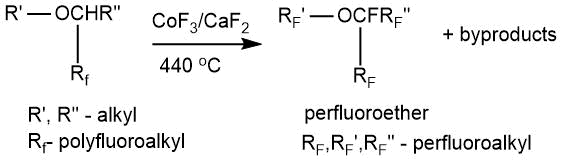
Фторирование проводили в горизонтальном никелевом реакторе с мешалкой при температуре 440°С смесью трифторида кобальта и дифторида кальция (1:1 масс.).
Так в результате фторирования 2,2,3,4,4,4-гексафтор-1-метилбутил этилового эфира был получен перфтор-1-метилбутил этиловый эфир с выходом 43%.
Фторирование 2,2,3,4,4,4-гексафторбутил метилового эфира приводило к получению перфторбутил метилового эфира с выходом 36%.
В работе Дж. К. Татлоу с соавт. [64] описано фторирование хлорсодержащих простых эфиров трифторидом кобальта при 110÷150°С. В результате фторирования 2-хлордиэтилового эфира была получена сложная смесь соединений, в которой были идентифицированы 2-хлор-1,1,2,2-тетрафторэтил 1,2,2-трифторэтиловый эфир, 2-хлор-1,2,2-трифторэтил 1,2,2,2-тетрафторэтиловый эфир, бис-1,2,2-трифтоэтиловый эфир и ряд других фторсодержащих эфиров.
5.3. Реакции спиртов с кетонами и карбоновыми кислотами
В статье японских исследователей Матсубара с соавт. [65] описан синтез 1Н,1Н,2Н,2Н-перфтороктил-1,3-диметилбутилового эфира, (F-626, выход 92%, т.кип. 214 °С), нагреванием (105°С, 8 ч) полифторалканола C6F13C2H4OH с кетоном СН3С(О)СН2СН(СН3)2 в токе водорода (180 мл/мин) в присутствии Pd/C с отгонкой воды в ходе реакции.
C6F13C2H4OH + CН3C(О)СH2CH(СH3)2 → C6F13C2H4OСН(СН3)СН2СН(СН3)2
Обычными продуктами взаимодействия спиртов, в том числе фторированных, с карбоновыми кислотами являются сложные эфиры. Однако, У. Хесс с соавт. [66] получили простой эфир при электролизе трифторэтанола с N-ацилглицином:
RC(O)NHCH2COOH + CF3CH2OH → RC(O)NHCH2OCH2CF3 + HCOOH,
R = CH3-, CF3-, C6H6-.
В работе Р.А.Беккера с соавт. [67] приведено описание получения неустойчивого перфтор-изо-пропенил метоксиметилового эфира взаимодействием хлорметилметилового эфира с хлормеркурпентафторацетоном (0°С, 50%).
Исходный хлормеркурпентафторацетон был синтезирован взаимодействием перфторпропен-2-ола с трифторацетатом ртути (20°С, 79,2%) с последующей обработкой ацетилхлоридом (-50°С, 97,9%).
CF2=C(OH)CF3 + (CF3COO)2Hg → CF3COOHgCF2C(O)CF3 → ClHgCF2C(O)CF3
ClHgCF2C(O)CF3 + ClCH2OCH3 → CF2=C(CF3)OCH2OCH3.
5.4. Реакции перфторированных нитрозоалканов со спиртами
Реакция перфторированных нитрозоалканов с гидроксиламином в спиртовых средах приводит к образованию соответствующих эфиров.
В статье А.В.Фокина и А.Т.Узуна [68] приведено описание синтеза алкилтрифторметиловых эфиров из трифторнитрозометана и гидроксиламина в среде спирта.
CF3NO + H2NOH + ROH → CF3OR
В цитируемой работе показано, что продуктами реакции 1-нитро-2-нитрозотетрафторэтана с гидроксиламином в метаноле являются 2-нитротетрафторэтилметиловый эфир (т.кип. 80-82°С) с выходом 26% и метиловый эфир дифторнитроуксусной кислоты (т.кип. 55°С/100 мм. Hg) с выходом 32%.
O2NCF2CF2NO + H2NOН + CH3OH → O2NCF2CF2OCH3 + O2NCF2C(O)OCH3
5.5 Взаимодействие спиртов c 2,4,6-трис-(2,2,2-трифторэтокси)-[1,3,5]триазином (TriTFET)
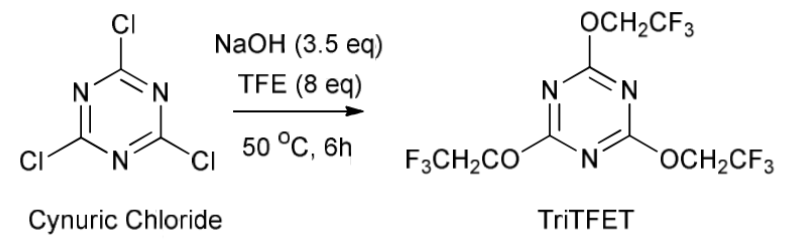
С.Мангава с сотр. [69] синтезировали трифторэтилбутиловый эфир взаимодействием н-бутанола с 2,4,6-трис-(2,2,2-трифторэтокси)-[1,3,5]триазином (TriTFET) в присутствии п-толуолсульфоновой кислоты в качестве катализатора. Фторалкилирующий реагент TriTFET был получен при обработке хлорангидрида циануровой кислоты (цианурхлорид) трифторэтанолом (TFE) в присутствии NaOH в качестве основания. Второй этап синтеза основан на взаимодействии н-бутанола с полученным на первом этапе фторированным алкилирующим реагентом, в условиях кислотного катализа.
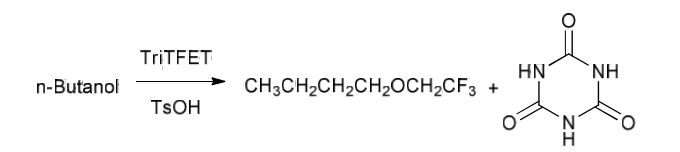
Было установлено, что оптимальными условиями синтеза фторированных эфиров являются использование 0.6 эквивалента TriTFET и ацетонитрила в качестве растворителя. При таких условиях синтеза выход эфиров достигает 94 %. Результаты представлены в Табл. 5.2.
Таблица 5.2. Условия синтеза фторированных эфиров [55]
|
No |
Растворитель |
TriTFET (экв.) |
Выход, % |
|
1 |
CH2Cl2 |
0.6 |
20 |
|
2 |
CH2Cl2 |
0.4 |
10 |
|
3 |
CH3CN |
0.6 |
94 |
|
4 |
CH3CN |
0.4 |
68 |
|
5 |
DMF1 |
0.6 |
78 |
|
6 |
DMF |
0.4 |
60 |
|
7 |
AсOEt2 |
0.6 |
52 |
|
8 |
AсOEt |
0.4 |
40 |
|
9 |
1,4-Диоксан |
0.6 |
84 |
|
10 |
1,4-Диоксан |
0.4 |
72 |
|
11 |
Толуол |
0.6 |
45 |
|
12 |
Толуол |
0.4 |
32 |
1Диметилформамид; 2этилацетат
Заключение
В обзоре предпринята попытка обобщения и систематизации существующих методов получения фторсодержащих простых эфиров. Авторы не ставили задачу исчерпывающего освещения всех источников и отдельных примеров синтеза. Тем не менее, авторы выражают надежду в том, что представленный материал может быть полезен в исследовательской деятельности.
Список литературы
- S.V. Vershilov, V.V. Kornilov at all, Fluorinated ethers. Communication 1//Fluorine Notes, Vol. 3(136), 2021, URL: http://en.notes.fluorine1.ru/public/2021/3_2021/article_1.htm; S.V. Vershilov, V.V. Kornilov at all, Fluorinated ethers. Communication 2//Fluorine Notes, Vol. 5(138), 2021, URL: http://en.notes.fluorine1.ru/public/2021/5_2021/article_4.html
- И.Л. Кнунянц, О.В. Кильдишева, И.П. Петров, О Взаимодействии алифатических окисей с фтористым водородом. I. // Ж. Общ. хим., 1949, Т. 19, Вып. 1, c. 95-100.
- M.L.Brey, P.Tarrant. The preparation and properties of some vinyl and glycidyl fluoroethers // J. Am. Chem. Soc., 1957, V. 79, No 24, pp. 6533-6536.
- Синтезы фторорганических соединений. (Мономеры и промежуточные продукты). // Под ред. акад. И.Л.Кнунянца и проф. Г.Г.Якобсона. М. Химия. 1977. 251 с.
- V. Grakauskas, Alkylatoin reactions of 2-fluoro-2,2-dinitroethanol // J. Org. Chem., 1970, v. 35, No. 9, pp. 3030-3036.
- А.В. Фокин, В.А.Комаров, А.Н. Воронков, В.И. Шевченко, Е.И. Любимова, Дифторнитроэтиловые эфиры 1,2-алкиленгликолей и их производных. // Изв. АН СССР, Сер. хим., 1975, c. 1448-1450.
- E.H. Banitt, Fluoroalkoxyalkyl-2-cianoacrylates and polymers thereof, Patent US 3532674, 1970.
- E.H. Banitt, Fluoroalkoxyalkyl-2-cyanoacetates, Patent US 3663592, 1972.
- R.W.-H. Chang, E.H. Banitt, R.W. Joos, Fluoroalkoxyalkyl-2-cianoacrylate compositions used in tooth treatment, Patent US 3540126, 1970.
- R.N. Thompson, M.F. Hoover, Fluorocarbon containing diallyl ammonium compound, Patent US 3717679, 1973.
- M. Hayek, R.J. Moody, Fluorocarbon-containing printing ink and process for image printing, Patent US 3948668, 1976.
- A.J. Szur, Nonionic fluorochemical surfactants, Patent US 3980715, 1976; Anionic fluorochemical surfactants, process of coating and treated polymeric shapes, Patent US 4208466, 1980.
- V. Grakauskas, Polynitroalkyl Ethers // J. Org. Chem. 1973, v. 38, No. 17, pp. 2999-3004.
- D.Sianesi, A. Pasetti, F. Tarli, The chemistry of hexafluoropropene epoxide // J. Org. Chem., 1966, V. 31, No 7, pp. 2312-2316.
- D.Sianesi, A. Pasetti, F. Tarli, Derivatives of fluorinated carboxylic acids and process for their preparation, Patent US 3535369, 1970.
- L .W.Breed, R.L Elliott., C.F. Key, Fluorinated Esters Lubricants Containing Ethers Structures // Ind. and Eng. Chem. Prod. Res. and Developm., 1972, V. 11, No 1, pp. 88-91.
- D.D. Coffman, M.S. Raasch, G,W. Rigby, P.L. Barrick, W.E. Hanford, Addition reactions of tetrafluoroethylene // J. Org. Chem., 1949, V. 14, pp. 747-753.
- E.T. McBee, W.F. Marzluff, O.R. Pierce, The Ionization Constants of some fluorine-containing alcohols // J. Am. Chem. Soc., 1952, V. 74, No. 2, pp. 444-446.
- V.Weinmayr. Hydrogen fluoride as a condensing agent. VI. Reactions of fluoroolefins with formaldehyde in hydrogen fluoride // J. Org. Chem., 1963, V. 28, pp. 492-494.
- Л.С.Герман, И.Л.Кнунянц. Реакции в безводном фтористом водороде. Конденсация формальдегида с галоидолефинами в HF // ЖВХО им. Д.И.Менделеева, 1966, Т. 11, Вып. 3, c. 354-355.
- Л.С.Герман, И.Л.Кнунянц. Реакции в безводном фтористом водороде. Синтез фторсодержащих простых и сложных эфиров // ЖВХО им. Д.И.Менделеева, 1966, Т. 11, Вып. 3, c. 356-358.
- V.Weinmayr, Process for preparing polyfuoro alkyl compounds, Patent US 2992276, 1961.
- H.L. Yale, Process for preparating halogenated propanols, Patent US 3415894, 1968.
- И.Л.Кнунянц, О.В.Кильдишева, Э. Быховская, Взаимодействие алифатических окисей с фтористым водородом. II // Ж. Общ. Хим., 1949, Т. 19, Вып. 1, c. 101-113.
- Gy.Olah, A.Pavlath, Synthesis of organic fluorine compounds. IV. Derivatives of 2-fluoroethanol of insecticidal effect // Acta Chim. Acad. Sci. Hung., 1954, V. 4, p. 89.
- G.C. Tesoro, R.N. Ring, Fluorinated compounds, Patent US 3420840, 1969.
- В.А.Комаров, С.М.Давыдова, К.В.Фросина, А.с. СССР 269156, БИ № 15. 23 (1970)
- В.А.Комаров, А.В.Фокин, К.В.Фросина, Х.А.Абдулганиева, Реакционная способность некоторых дифторнитросодержащих спиртов // ЖОХ, 1967, Т. 38, Вып. 3, c. 684-686.
- H.G. Adolph, M.J. Kamlet, Fluoronitroalkanes. IV. Some reactions of 2-fluoro-2,2-dinitroethanol // J. Org. Chem. 1969. V. 34. N. 1. pp. 45-50.
- M.E. Hill, K.G. Shipp, Process for acetal preparation, Patent US 3526667, 1970.
- А.Я.Запевалов, Е.О.Землякова, А.В.Пестов, А.М.Семенова, Способ получения бис(2,2,3,3,4,4,5,5-октафторпентилокси)метана, Патент RU 2747026, 2021.
- Л.С. Богуславская, В.С. Этлис, К.В. Яровых, А.Б. Буловятова, Сопряженное галоидирование непредельных соединений. IV. Хлорфторирование 2-галоидаллиловых спиртов и некоторые реакции фторхлорпропанолов // Ж. Орг. хим., 1971, Т. 7. Вып. 7, c. 1338-1343.
- M.E. Hill, L.O. Ross, Reduction of difluoronitroacetate esters. The preparation and properties of 2,2,2-difluoronitroethanol and novel formation of hemiacetals by reduction // J. Org. Chem., 1967. V. 32. N. 8. pp. 2595-2600.
- A.H. Ahlbrecht, Fluorinated Ethers, Patent US 3818074A, 1974.
- D.W. Codding, Fluorocarbon vinyl ethers and polymers, Patent US 2732370, 1956.
- H.G.Adolph, 2,2-Dinitroalkyl vinyl ethers and polymer thereof, Patent US 3808182, 1974.
- L.S. Croix, Preparation of 2,2,2-trifluoroethyl vinyl ether, Patent USA 2872487, 1959.
- В.С. Сухинин, С.И. Минеев, Синтез и полимеризация виниловых эфиров теломерных спиртов // ЖВХО им. Д.И.Менделеева, 1981, Т. 26, № 3, c. 344-345.
- B.K. Mandal, Z. Yue, X. Mei, H. Dunya, Q. Ma, Synthesis and physical properties of new fluoroether sulfones // J. Fluor. Chem., 2018, V. 216, pp. 118-123.
- V. Tortelli, I. Wlassics, C. Monzani, B.L.Kent, A.Vesteroni. Hydro-fluorocompounds, WO 2012160135, 2012.
- Z. Yue, H. Dunya, S. Aryal, C.U. Segre, B. Mandal, Synthesis and electrochemical properties of partially fluorinated ethers solvents for lithium-sulfur battery electrolytes // J. Power Sources, 2018, No 401, pp. 271-277.
- B. Boutevin, B. Youssef, Synthese d’ethers vinyliques a chaine laterale fluoree // J. Fluorine Chem., 1989, Vol. 44, pp. 395-412.
- G. Binch, E.L. Eliet, S. Mager, Ring inversion barrier in 5,5-difluoro-1,3-dioxane // J. Org. Chem., 1973, V. 38, No 23, pp. 4079-4081.
- К.А. Петров, Н.А. Тихонова, Н.А. Щекотихина, Реакции пергалоидированных карбонильных соединений и их гидратов с ортоэфирами и ацеталем ацетальдегида // Ж. орг. хим., 1977, Т. 13, № 5, c. 943-948.
- J.H. Simons, The electrochemical process for the production of fluorocarbons // J. Electrochem Soc., 1949, 95, pp. 47–66.
- J.H.Simons. Fluorocarbon ethers. Patent US 2500388, 1950.
- S.Benninger, T.Martini, S.Rebsdat. Aliphatic and cyclic perfluoro-alkyl ethers and process for the preparation thereof, Pat. US 3962348, 1976.
- S. Nagase, H. Baba, R. Kojima, Preparation of Perfluorocarboxylic Acids from Unsaturated Compounds by Electrochemical Fluorination // The Journal of the Society of Chemical Industry Japan, 1962, Vol.65, Iss.1, p. 1183, DOI: 10.1246/nikkashi1898.65.38.
- K. Okazaki, S. Nagase, H. Baba, K. Kodaira, Electrochemical fluorination of chlorine-containing ethers // J. Fluor. Chem., 1974, Vol. 4, p. 383.
- J.C.Hansen, Preparation of F-alkyl F-isobutyl ethers by electrochemical fluorination, Patent US 5474657, 1995.
- Г.И.Каурова, Электрохимическое фторирование органических соединений // Химическая промышленность, 2017, Т. XCIV, Вып. 4, с. 163-207.
- В.М. Маталин, Синтез перфторированных органических соединений методом электрохимического фторирования в присутствии третичных аминов // Дисс. на соискание ученой степени к.х.н., СПб, 2008.
- V.G. Barabanov, V.A. Matalin, G.I. Kaurova, D.D. Moldavskij, Perfluorinated organic compound production process, Patent RU 2221765, 2004.
- M. Napoli, A. Scipioni, G. P. Gambaretto, F.M. Carlini, M. Bertola, Yield improvement in the electrochemical production of perfluoro-octanoic acid // J. Fluorine Chemistry, 1994, V. 67, pp. 261-264.
- T. Abe, E. Hayashi, H. Baba, K. Kodaira, S. Nagase, Fluorination of 2-alkyl-substituted oxanes. The synthesis and purification of perfluoro(2-alkyl-substituted oxane)s // J. Fluorine Chem., 1980, V. 15, pp. 353-380.
- H. Ishii, Y. Hou, T. Fuchigami, Electrolytic Partial Fluorination of Organic Compounds. Part 41. Highly Selective Electrolytic Fluorination of Dimethoxyethane, its Homologues, and Crown Ethers // Tetrahedron, 2000, V. 56, p. 8877.
- H. Muramatsu, K. Inukai, T. Ueda, The Radiation-Induced Addition Reaction of Ethers to Chlorofluoroolefins // Journal of Organic Chemistry, 1964, V. 29, Iss. 8, p. 2220-2223.
- H.Muramatsu. H.Kimoto, K.Inukai, The addition reactions of fluoroalkyl ethyl ethers to perfluoropropene // Bull. Chem. Soc. Japan, 1969, V. 42, No 4, pp. 1155-1158.
- R. Chambers, B. Grievson, N. Kelly, Free radical chemistry. Part 3. Substituent Effects in Additions of Ethers to Fluorinated Alkenes // J. Chem. Soc. Perkin Trans. I, 1985, pp. 2209-2213.
- J.L. Adcock, R.A. Beh, R.J. Lagow, Successful direct fluorination of oxygen-containing hydrocarbons // Journal of Organic Chemistry, 1975, Vol. 40, pp. 3271-3275, https://doi.org/10.1021/jo00910a024.
- M. Stacey, J. C. Tatlow and A. G. Sharpe (Eds.): Advances in Fluorine Chemistry. London: Butterworths 1960 (vol. 1), 1961 (vol. 2), 1963 (vol. 3).
- M. Brandwood, P.L. Coe, C.S. Ely, J. C. Tatlow, Polyfluoro diethyl and ethyl methyl ethers: their preparation using cobalt (III) fluoride and potassium tetrafluorocobaltate (III) and their dehydrofluorination // Journal of Fluorine Chemistry, 1975, Vol. 5, pp. 521-535.
- R. Chambers, B. Grievson, Free radical chemistry. Part 5. A new approach to the synthesis of perfluorinated ethers // Journal of Fluorine Chemistry, 1985, Vol. 29, pp. 323-339.
- P. L. Coe, M. S. Lennard, J. C. Tatlow, Chloropolyfluorodiethyl ethers // Journal of Fluorine Chemistry, 1996, Vol. 80, pp. 87-90.
- H. Matsubara, S. Yasuda, H. Sugiyama, I. Ryu, Y. Fujii, K. Kita, A new fluorous/organic amphiphilic ether solvent, F-626: execution of fluorous and high temperature classical reactions with convenient biphase workup to separate product from high boiling solvent // Tetrahedron, 2002, V. 58, pp. 4071-4076.
- U. Hess, T. Gross, R.Thiele, Zur Kolbe-Reaction von Aminosauren // Zeitschrift fur Chemie, 1979, 19, N. 5, pp. 195-196.
- А.В. Фокин, А.Т. Узун, Исследование реакционной способности 1-нитро-2-нитрозотетрафторэтилена. I. Взаимoдействие с гидроксиламином // Ж. общ. хим. 1966. Т. 36. № 1. c. 117-119.
- S.K. Mangawa, C. Sharma, A.K. Singh, S.K. Awasthi, Expedient and efficient one pot synthesis of trifluoroethyl ethers from metal free 2,4,6-tris(2,2,2-trifluoro-ethoxy)-[1,3,5] triazene // The Royal Society of Chemistry, 2012, V. 1, pp. 1-5.
Статья рекомендована к публикации членом редколлегии д.х.н. С.М. Игумновым
Fluorine Notes, 2022, 140, 1-2


