Поступило в редакцию: Ноябрь 2021
УДК 542.91:547.416
Fluorine Notes, 2021, 139, 3-4
<α-ФТОРАЛКИЛАМИНЫ, ИХ СВОЙСТВА И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИСТОЧНИКОВ НУКЛЕОФИЛЬНОГО ФТОР-ИОНА
В.Э. Бойкоа,б, В.Л. Дона,б
аИнститут элементоорганических соединений им. А. Н. Несмеянова РАН,119991, Москва, ул. Вавилова, д. 28.
e-mail: boykii@mail.ru
бООО НПО «ПиМ-Инвест», 119991, Москва, Ленинский проспект, 47.
Аннотация: Мини-обзор посвящен α-фторалкиламинам, в том числе соединениям, содержащим более одного атома азота в α-положении к фтору, их получению, свойствам и применению в синтезе фторалифатических и фторароматических соединений в качестве источников нуклеофильного фтор-иона.
Ключевые слова: фтор-ион, α-фторалкиламины, фторметилдиметиламин, дифторметилдиметиламин, трифторметилдиметиламин, бис-(диметиламино)дифторметан, формамидиний гидрофторид, гуанидиний фторид, трис-(диметиламино)фторметан.
Фтор-ион играет большую роль в химии фторорганических соединений. Он не только способен замещать другие галогены и различные уходящие группы на фтор, но, что особенно важно, способен генерировать фторсодержащие анионы из электрофильных ненасыщенных соединений. Нуклеофильность фтор-иона кардинальным образом зависит от среды. В протонных растворителях, а особенно в воде, фтор-ион в силу своего малого радиуса и высокой плотности заряда образует сильные водородные связи и вследствие этого является слабым нуклеофилом. Внутренняя сфера гидратированного фтор-иона содержит пять прочно связанных молекул воды, в отличие от ионов хлора (три) брома (две). [1]. Этим объясняется порядок нуклеофильности галогенид ионов в воде I- < Br- < Cl- >> F- [2, 3]. В полярных апротонных растворителях порядок силы нуклеофила меняется на обратный: F- >>Cl- > Br- > I- [1]. В полярных апротонных растворителях нуклеофильность фтор-иона зависит в том числе и от катиона. Зависимость обусловлена преимущественно радиусом катиона, определяющим способность к диссоциации [4, 5]. Источники фтор-иона для большинства реакций это в первую очередь фториды щелочных металлов. Растворимость неорганических фторидов в полярных апротонных растворителях мала. Увеличивает нуклеофильность фтор-иона специфическая сольватация катиона, как например комплексообразование с краун эфирами [6]. Значительно больше растворимость тетраалкиламмониевых солей, и они являются очень эффективными источниками фтор-иона для многих реакций, однако их чрезвычайно сложно получить в безводном виде, а наличие воды снижает активность фтор-иона [7]. Из экономических соображений, для масштабных промышленных синтезов в 70-80 годы этот выбор ограничивался фторидом калия. Задача поиска новых альтернативных источников фтор-иона была крайне актуальна в 70-80 годы прошлого века, когда И. Л. Кнунянц обратил свое внимание на α-фторалкиламины, и продолжает оставаться актуальной сейчас.
1. α-Фторалкиламины
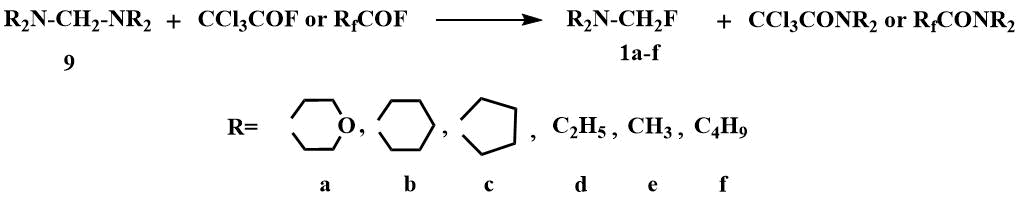
Известен целый ряд α-фторалкиламинов, к которым относятся фторметилдиалкиламины, в том числе фторметилдиметиламин (1) [8], дифторметилдиметиламин (2) [9-10], трифторметилдиалкиламины (3) [11-14],
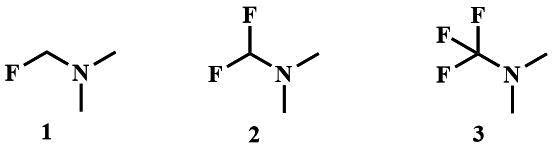
полифторалкиламины - наиболее изученные из них, получаемые взаимодействием фторолефинов с диалкиламинами, которые называют иначе фторалкиламино реагенты (ФАР) (4a-c) [15-17], а также перфтортриалкиламины (5) [18-22].

Подобно остальным третичным аминам α-фторалкиламины, за исключением перфторалкиламинов, лишенных основных свойств [23], образуют четвертичные аммониевые соли при действии алкилирующих агентов. Кристаллический йодистый фторметил(триметил)аммоний образуется при смешении фторметилдиметиламина (1) с йодистым метилом [8], такая же соль получена из дифторметилтриметиламина (2) [24].
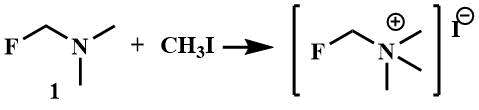
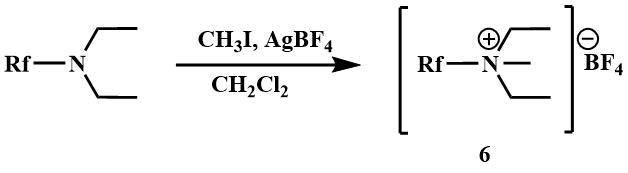
Четвертичные соли трифтортриметиламина и полифтортриалкиламинов (6) получены взаимодействием с йодистым метилом в присутствии тетрафторбората серебра [23].
Общим свойством α-фторалкиламинов является высокая подвижность α-атомов фтора. Причиной подвижности α-атомов фтора является стабилизация катиона, образующегося при отрыве фтор-иона, неподеленной парой азота [25].
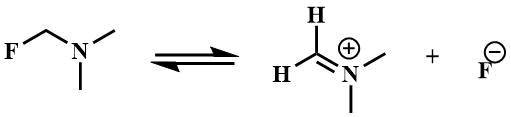
Все α-фторалкиламины легко гидролизуются водой и энергично взаимодействуют с различными гидроксилсодержащими соединениями. Это их свойство нашло широкое применение для замещения гидроксильных групп на фтор.
Наиболее широко для этой цели применяются ФАР: реагенты Яровенко (4а) и Ишикавы (4b), получаемые соответственно взаимодействием хлортрифторэтилена и перфторпропилена с диэтиламином, а также продукт взаимодействия с диэтиламином тетрафторэтилена, называемый также реагентом Петрова (4с).
Из спиртов взаимодействием с ФАР получают монофторалканы [15-17, 28], а из кислот и из сульфокислот - фторангидриды и сульфонилфториды [15, 26-28]. Вторым продуктом этой реакции является соответствующий амид (7), в случае реактива Яровенко - диэтиламид хлорфторуксусной кислоты, Ишикавы - диэтиламид 2,3,3,3-тетрафторпропионовой кислоты, и диэтиламид дифторусксусной кислоты в случае реагента Петрова.
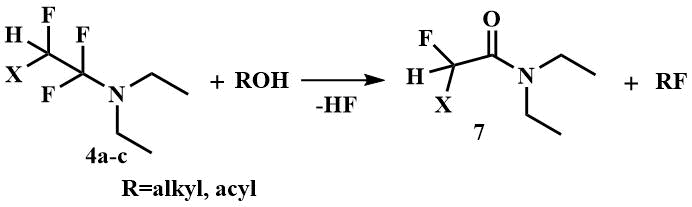
С кислотами Льюиса фторалкиламины, в частности ФАР, образуют иминиевые соли (8)
[29].
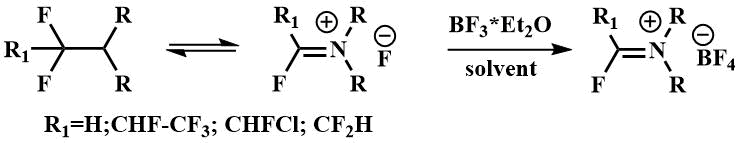
Такие иминиевые соли используют в синтезе фторсодержащих гетероциклов [29-31].
Реакциям ФАР, наиболее часто используемых в качестве фторирующих реагентов для замены гидроксила на фтор: реактивам Яровенко, Ишикавы и Петрова, а также применению полученных из них взаимодействием с кислотами Льюиса иминиевых (иммониевых) ионов в синтезе гетероциклов посвящен ряд обзоров [30-38].
Здесь нам хотелось бы подробнее остановиться на других α-фторалкиламинах, как ковалентных содинениях, которые однако могут рассматриваться как равновесные мезомерные иминиевые структуры, способные выступать в качестве источников фтор-иона, так и ионных бис- и трис-(диамино)фторметанах, где фтор-ион координирован с алкилиминиевым катионом, которые также представляют огромный интерес в качестве источников фтор-иона.
1.1. Фторметилдиметиламин
Фторметилдиалкиламины были впервые получены в 1970 г. расщеплением соответствующих аминалей (9) фторангидридами карбоновых кислот [8].
В отличие от полученных ранее хлорметил-, брометил- и йодметилдиалкиламинов, которые проявляли солеобразные свойства и рассматривались как карбиминиевые соединения [39], фторметилдиалкиламины оказались жидкостями с невысокой температурой кипения, растворимыми в полярных и неполярных органических растворителях, типичными ковалентными соединениями [8]. Однако в 19F спектре фторметилдиалкилмина (1b) спин-спиновое взаимодействие F-H наблюдалось только при отрицательных температурах, при повышении температуры оно исчезало, во фторном спектре триплет переходил в синглет, а, соответственно, в протонном спектре дублет в синглет, что свидетельствовало о быстром обмене фторов между молекулами, т.е. о преобладании ионной структуры [8].
Простейший из фторметилдиалкиламинов фторметилдиметиламин представляет собой бесцветную жидкость, кипящую 46С при нормальном давлении, растворимую в пентане, эфире и других полярных и неполярных органических растворителях. В начале двухтысячных фторметилдиметиламин явился объектом исследований молекулярной структуры и квантово-химических расчетов, была построена модель молекулы в газовой фазе [25].
Исследование FCH2NMe2 газовой электронографией показало его существование в виде единственного конформера с антиперепланарной ориентацией C–F связи по отношению к неподеленной паре N (см. Рис.1).
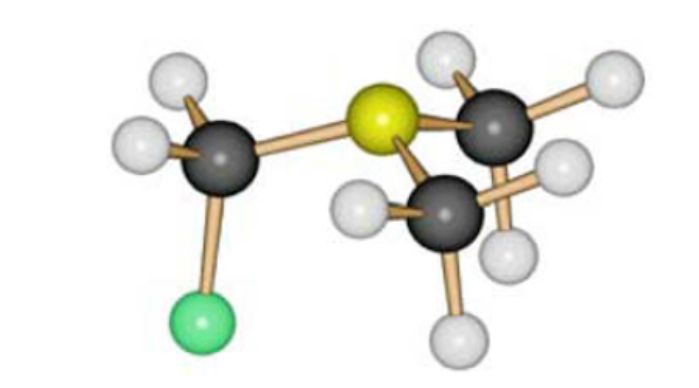
Рисунок 1. Молекулярная модель FCH2NMe2.
Донорно-акцепторное взаимодействие между неподеленной электронной парой и вицинальными σ* орбиталями - так называемое аномерное взаимодействие, рассматриваемое в классическом варианте как резонансные структуры амин - ион иминия, приводит к ожидаемым структурным последствиям - укорочению связи между атомом азота и связанным со фтором атомом углерода [N–CF = 1,408Å], по сравнению с длиной связи азот – метильная группа [N–CMe = 1,466Å], удлинению связи C–F до 1,410Å и увеличению углов связей азота и углерода. Рассчитанные квантово-химическим путем величины близки к экпериментальным. Рассчитаная квантово-химическим путем энергия взаимодействия неподеленной пары азота с σ* орбиталью С–F связи в антиперепланарной ориентации составляет 28,2 ккал/моль [25].
Эти исследования хорошо согласуются с открытыми в 80-х годах в лаборатории Кнунянца реакциями.
Исследуя химические свойства фторметилдиметиламина, в лаборатории Кнунянца обнаружили, что он легко присоединяется к перфторолефинам - перфторпропилену, перфторизобутилену по ионному механизму [40].
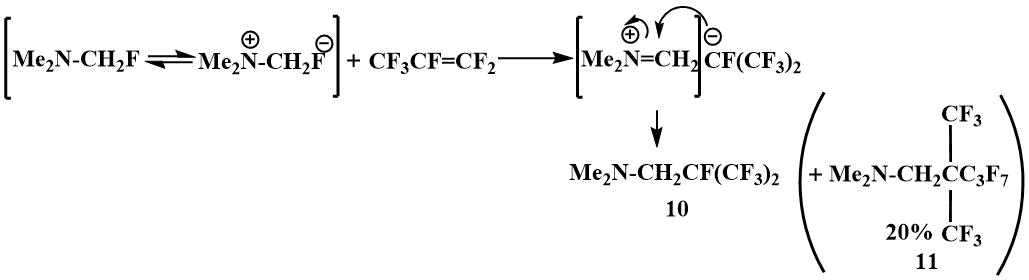
Ионный механизм этой реакции подтверждается образованием характерных побочных продуктов. Для образования (11), которого получилось 20% в результате реакции с перфторпропиленом необходимо, чтобы вначале образовался димер перфторпропилена, образование которого инициируется F-ионом [40].
Подобным образом фторметилдиметиламин реагирует с перфторизобутиленом, давая перфтор(трет-бутил)триметиламин Me2NCH2C(CF3)3, который однако, легко гидролизуется соляной кислотой, в отличие от продукта взаимодействия с перфторпропиленом - перфторизопропилтриметиламина (10), образующего устойчивый хлоргидрат.
Реакция с перфторазапропеном (12), который реагирует исключительно как электрофил, также, по-видимому, начинается с отрыва фтор-иона из фторметилдиметиламина [40].
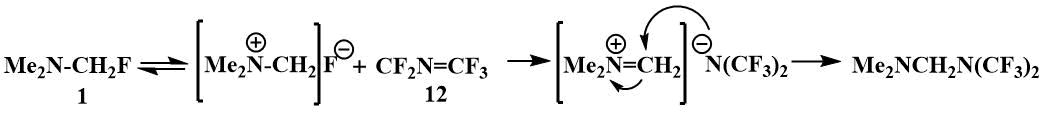
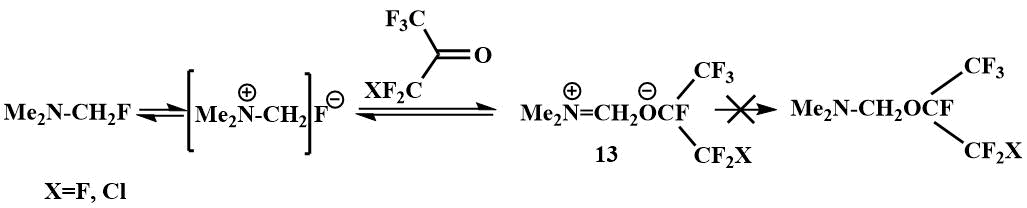
Фторметилдиметиламин реагирует и с фторкетонами, но при этом образуются ионные соединения – алкоголяты N,N-диметилметилениминия (13), ионная структура которых подтверждена их физическими свойствами и спектральными данными. Эти соли хорошо растворяются в полярных растворителях (ДМФА, ацетонитрил), но не растворяются в эфире. Некоторые данные говорят об обратимости процесса присоединения фторметилдиметиламина к фторкетонам. Сигналы одиночного F в спектре соли (13) пропадают выше -20°С, что свидетельствует об обменных процессах, связанных, по-видимому, с обратимостью присоединения фторметилдиметиламина к полифторкетонам. [41].
Эти соли легко реагируют с перфторпропиленом и перфторизобутиленом, образуя продукты присоединения перфторалкокси-аниона и метилениминиевого катиона по кратной связи олефина. Поскольку легкость присоединения уменьшается от перфторизобутилена к перфторпропилену и тетрафторэтилену, который совсем не удалось вовлечь в эту реакцию, авторы предположили нуклеофильный механизм присоединения соли, когда сначала происходит атака перфторалксианиона на олефин, а затем полученный β-алкоксикарбанион ковалентно связывается с метилениминиевым катионом, образуя перфтор(алкоксиалкил)амин (14).
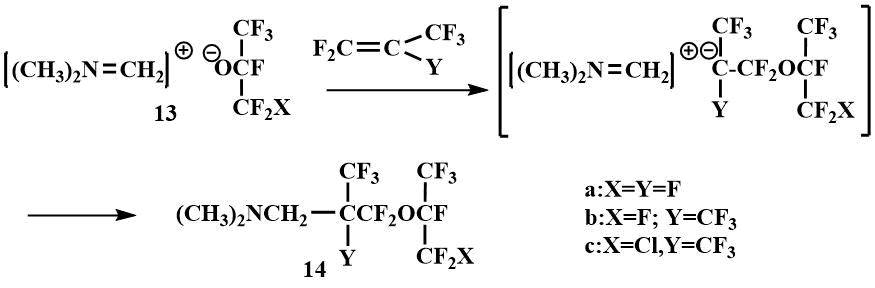
В пользу обратимости процесса присоединения фторметилдиметиламина к кетонам говорит и образование Me2NCH2C(CF3)3 в качестве побочного продукта наряду с (14) при присоединении соли (13) к перфторизобутилену при температуре выше -10°С [41].
Образование диалкиламинофторметанов, способных генерировать фтор-ион, при взаимодействии тетраалкилдиаминометанов - бис-(диметиламино)метана, бис-(диэтиламино)метана, бис-морфолино- и бис-пиперидинометанов с фторангидридами перфторкарбоновых кислот, дало возможность использовать тетраалкилдиаминометаны в качестве катализаторов конденсации окиси перфторпропилена с фторангидридами перфторкарбоновых кислот с целью получения фторангидридов перфторалкоксипропановых кислот (16) - исходных для получения перфторалкоксивиниловых мономеров - сырья для получения полимеров, обладающих специальными свойствами [42].
Конденсация в присутствии каталитического количества N,N,N',N'-тетраалкилдиаминометанов протекает с промежуточным образованием диалкиламинофторметана, который генерирует F-ион, инициирующий образование перфторалкоксианиона (15) взаимодействием с фторангидридом перфторкарбоновой кислоты.
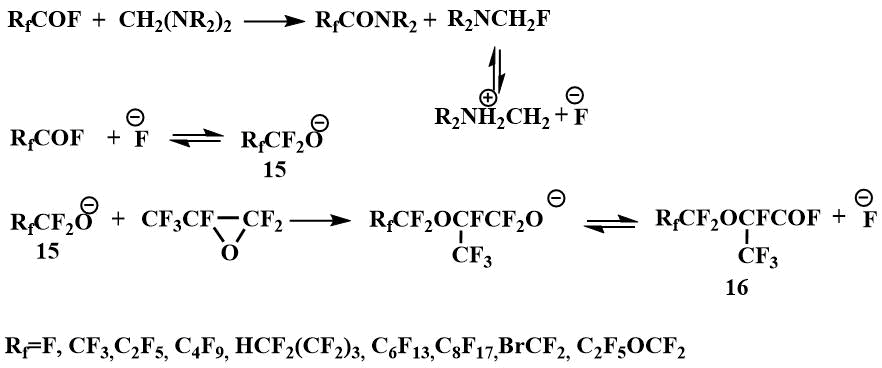
В дальнейшем такие реакции легли в основу производства, запущенного С.М. Игумновым в Пермском отделении Российского Центра Прикладной Химии (Пермский филиал ГИПХ) - получения перфторвиниловых эфиров, в частности перфторметилвинилового (17) и перфторбромэтилвинилового (18), и поверхностно активных материалов на основе перфторалкоксипропановых кислот (19) [43]. Для этих процессов использовался N,N,N',N'-тетраэтилметилендиамин в каталитических количествах. Применение этого каталитического метода позволило проводить многие процессы с участием О-анионов в температурном интервале от -10°С до комнатной и при атмосферном давлении, в то время как использование иных источников F-иона в аналогичных процессах требует повышенных температур (90- 150°C) и давления (5 - 200 атм) [44].
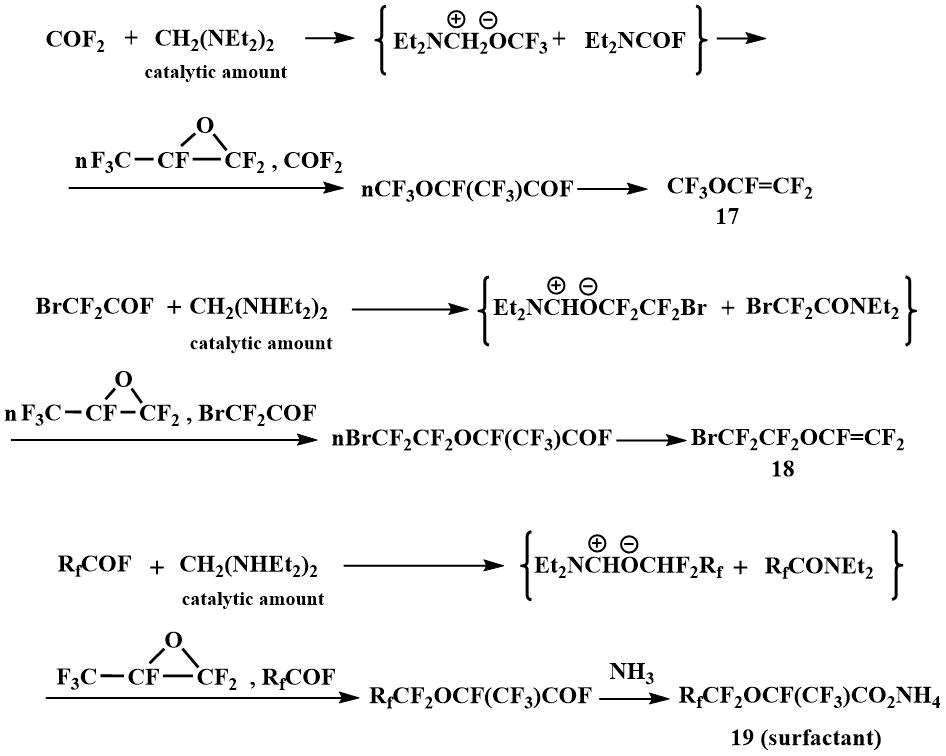
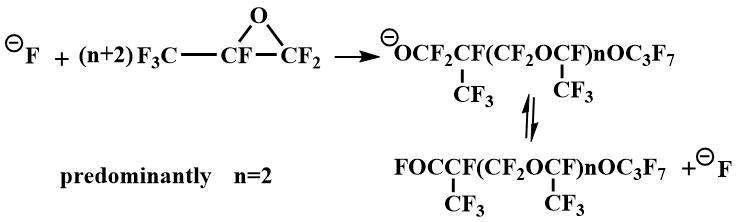
Тетраалкилдиаминометаны также были использованы в качестве катализаторов олигомеризации окиси перфторпропилена, при катализе тетраалкилдиаминометанами преимущественно получается тетрамер окиси.
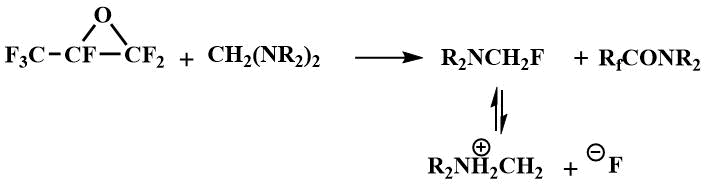
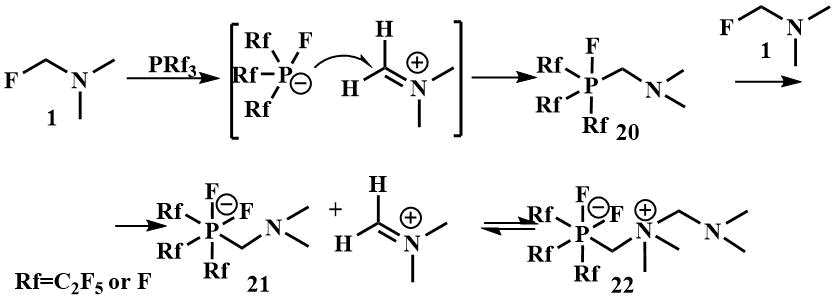
Пример взаимодействия фторметил(диметил)амина с фосфорными электрофилами изучен Рошенталлером и Хоге,
в результате взаимодействия фтортриметиламина с трифторидом фосфора или триалкилфосфинами получается
твердое вещество, представляющее собой цвиттер-ион (22). Предполагаемый механизм
взаимодействия включает первоначальный перенос фтор-иона от фторметилдиметиламина к PRf3 с образованием аниона [Rf3PF]- и иминиевого катиона, атаку этим анионом
атома углерода иминиевого катиона с образованием фосфорана Rf3PFCH2NMe2 (20),
далее отрыв фосфораном фтор-иона из второй молекулы Me2NCH2F с образованием
фосфата (21), который координируется с оставшимся иминиевым ионом, образуя цвиттерион (22) [45].
Обработка продукта реакции с P(C2F5)3 – [(C2F5)3PF2(CH2NMe2CH2NMe2)] (18) водным гидроксидом натрия приводит к образованию перфторалкилфторфосфата натрия - Na+ [(C2F5)3PF2 (CH2NMe2)]ˉ (19) [45].
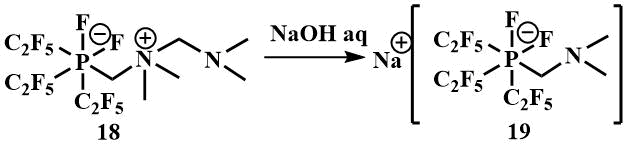
Перфторалкилфторфосфаты щелочных металлов находят применение в электронной промышленности, выгодно отличаясь от традиционно применяемых гексафторфосфатов большей термостабильностью и устойчивостью к гидролизу, и в качестве ионных жидкостей [46].
1.2. Дифторметилдиметиламин
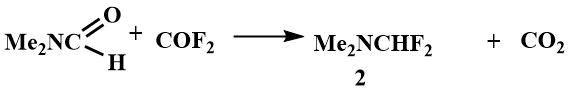
Дифторметилдиметиламин (2), полученный впервые в 1962 г. [2] взаимодействием карбонилфторида с диметилформамидом, представляет собой легкокипящую жидкость (т.кип. 49-51°С) , в его спектре и при 20°С наблюдается спин-спиновое взаимодействие между атомами фтора и водорода, что определенно указывает на ковалентный характер связи, с то время как аналогичное хлорное производное имеет чисто ионный характер [(CH3)2N=CHCI]+Cl–, и представляет собой гигроскопичную нелетучую соль [47].
Рентгенографическое исследование четвертичной соли (CH3)3NCHF2, проведенное в 1986 году, показало, что длина связи N-CHF2 (1,497 Å) меньше, чем связи N-CH3 (1,508 Å) [24].
Дифторметилдиметиламин также проявляет химические свойства, обусловленные подвижностью атомов фтора - экзотермически разлагается водой, при взаимодействии с трехфтористым бором образует тетрафторборат N,N-диметилфторметилениминия, реагирует с карбоновыми кислотами с образованием фторангидридов при 0С и с бензальдегидом, давая α,α-дифтортолуол с хорошим выходом [10].
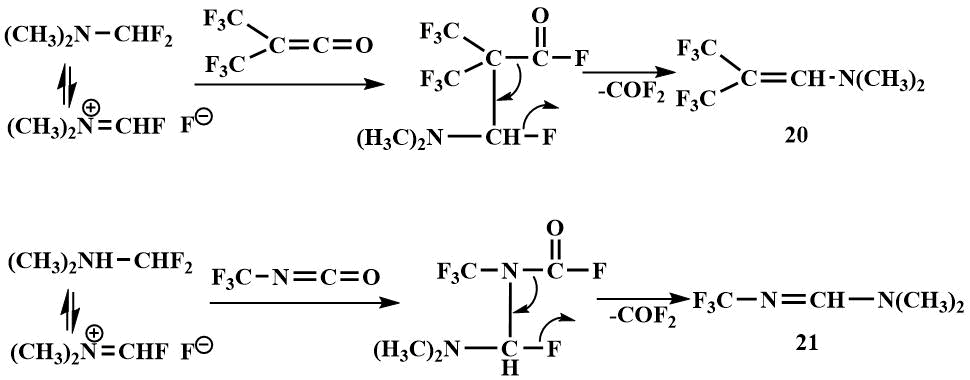
Дифторметилдиметиламин, как и фторметилдиметиламин, как было показано, способен быть источником фтор-иона при взаимодействии с сильно электрофильными соединениями, такими как бистрифторметилкетен и трифторметилизоцианат, давая гексафторизобутенилдиметиламин (20) и N-трифторметил-N',N'-диметилформамидин (21). Предполагается, что первоначально амин присоединяется по кратной связи ненасыщенного соединения, затем промежуточные продукты подвергаются дефосгенированию [48].
При взаимодействии с гексафторацетоном образуется нерастворимый в эфире кристаллический продукт, по аналогии с реакцией фторметилдиметиламина можно предположить образование соли (22), однако его не удалось идентифицировать, поскольку при температуре выше -30°С он разлагается, предположительно через диметиламинофторкарбен (23), который с избытком гексафторацетона образует диоксолан (24) (Был выделен и идентифицирован продукт гидролиза диоксолана) [48].
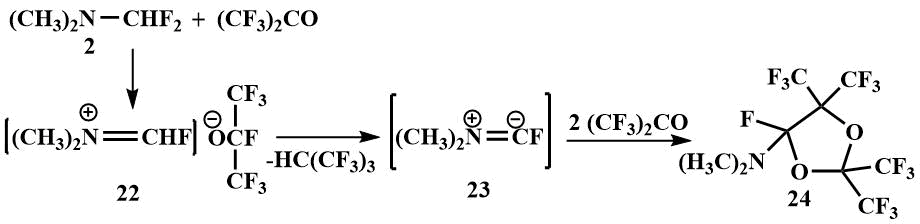
Дифторметилдиметиламин (2) присоединяется к перфторизобутилену, образуя иминиевую соль, стабилизированную третбутиланионом (25), которая находится в равновесии с солью (2а). При 20°C третбутил-анион атакует углерод катиона иминиевой соли (25) и образуется 1-фтор-1-перфтор(трет-бутил)триметиламин (26), а при 80°C третбутиланион соли (25) депротонирует катион с образованием гидроперфторизобутана и диметиламинофторкарбена (23), который реагирует с перфторизобутиленом, частично находящемся в равновесии (2а) ↔ (25), образуя 1-диметиламино-перфтор((3-метил)бутен-2) (27) с выходом 80% [48].
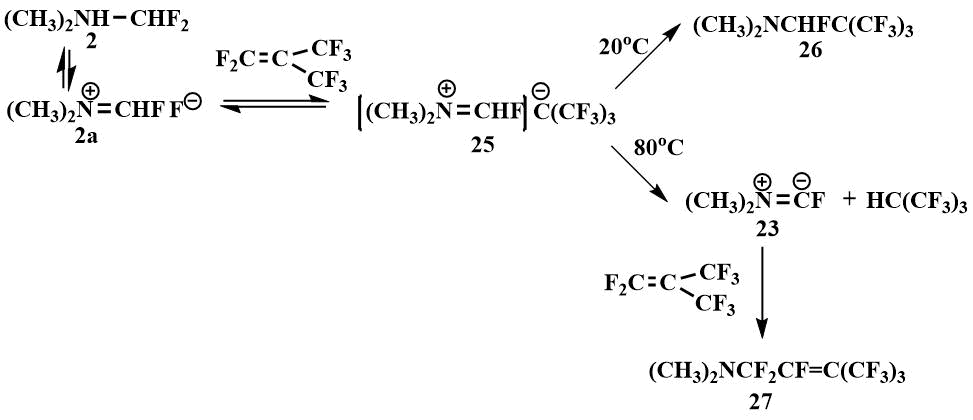
В последнее время возник интерес к применению дифторметилдиметиламина в качестве фторирующего агента. Недавно дифторметилдиметиламин был с успехом применен как фторирующий агент для замены гидроксила на F в синтезе интермедиата (28) для лекарственного препарата фторфеникола. Фторирование проводили в дихлорметане, авторы сообщают, что применение дифтордиметиламина позволило сократить отходы, поскольку побочный продукт (диметилформамид) используют повторно для получения дифтордиметиламина, снизить температуру, необходимую для реакции, по сравнению с фторированием реагентом Ишикавы, и повысить выход до 97% [49].
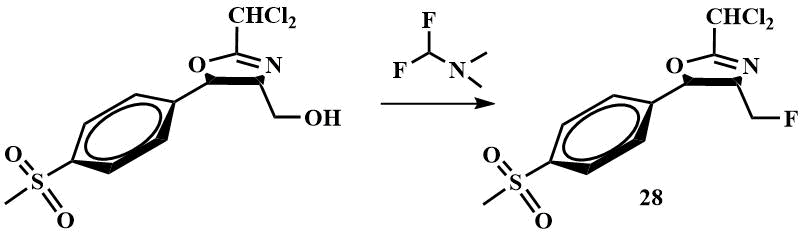
Другие авторы сообщают, что с его помощью возможно проводить фторирование вторичных спиртов. 1-Фторэтилбензол получен из соответствующего спирта с выходом более 80% [50].
1.3. Трифторметилдиметиламин
Трифторметилдиалкиламины, впервые полученные в 1957 г, представляют собой ковалентные соединения [51].
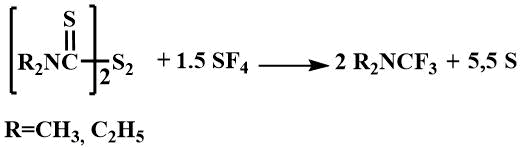
Трифторметилдиметиламин представляет собой легкокипящую жидкость с температурой кипения 20°C, трифторметилдиэтиламин кипит 71°C.
Эти соединения были получены также фторированием диметилформамида четырехфтористой серой в присутствии фтористого калия [14] или фторированием соответствующего трихлорметилдиалкиламина трехфтористой сурьмой [52].
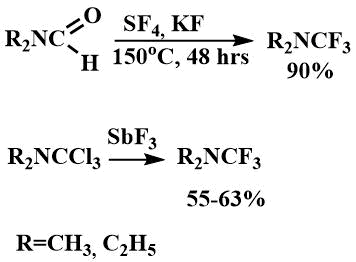
Рентгенографическое исследование четвертичной соли трифторметилдиметиламмоний иодида, проведенное в 1986 г. показало, что разница межатомных расстояний N-CF3(1,491Å) и N-CH3 (1,514Å) становится еще существеннее, чем для соответствующей соли дифторметилдиметиламина [24].
Трифторметил(диалкил)амины весьма реакционноспособные соединения, дымят на воздухе, бурно разлагаются водой, давая соответствующие формиламины (29).
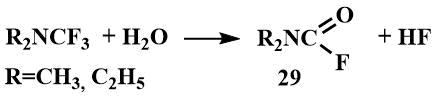
Дмовский с сотрудниками исследовали возможность применения трифторметилдиэтиламина в качестве фторирующего агента, и пришли к выводу, что он может быть полезен для фторирования вторичных и третичных спиртов, в то время как взаимодействие с первичными спиртами дает сложные смеси продуктов. [53]. Так при фторировании им изопропанола изопропилфторид получен с выходом 75%, а при фторировании трет-бутанола получается 3,5:1 трет-бутилфторида и 2-метилпропена, в то время как при использовании реактива Яровенко побочный продукт преобладает [53].
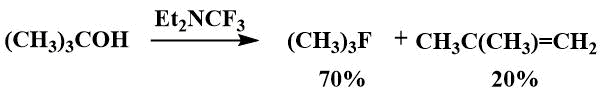
Трифторметилдиэтиламин не удалось присоединить к перфторолефинам, кроме того он не катализирует олигомеризацию гексафторпропена даже при повышенных температурах [53].
2. Соединения, содержащие более одного атома азота в α-положении к фтору
2.1. N,N,N',N'-формамидиний бифторид
Известно, что стабильность иминиевых катионов увеличивается в ряду
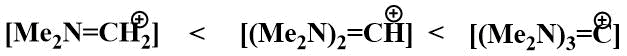
В поисках источника фтор-иона для того, чтобы повысить устойчивость иминиевого катиона, а значит и его способность генерировать карбанионы, в лаборатории Кнунянца в 80-е годы были предприняты попытки получения соединений, содержащих более одного атома азота в α-положении к фтору.
Первой целью исследований стало получение бис-(диметиламино)фторметана. Замещение подвижного атом фтора в дифторметилдиметиламине на диметиламино группу идет уже при комнатной температуре, однако, бис-(диметиламино)фторметан, образующийся в реакции дифторметилдиметиламина с диметиламином, удалось выделить только в виде фторгидрата, бифторида тертаметилформамидиния (ТМФБФ) (30), обладающего ярко выраженным ионным характером.
По-видимому, фтор-ион образующегося бис-(диметиламино)фторметана отрывает HF от фторгидрата диметиламина, что и приводит к образованию соли (30).
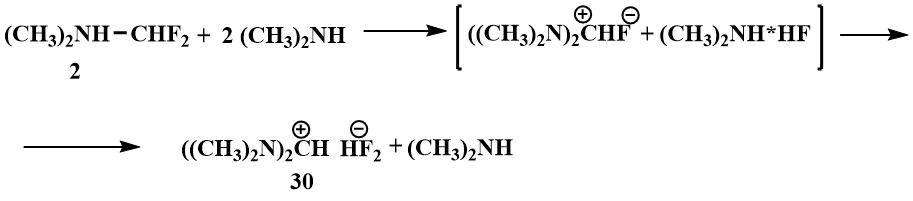
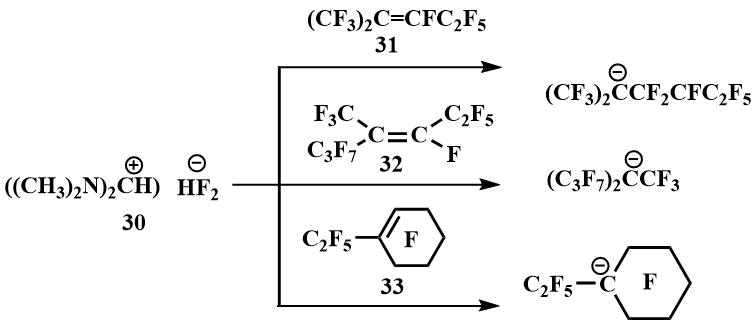
В присутствии бифторида ТМФБФ (30) Игумнову удалось получить и зафиксировать методом ЯМР при пониженных температурах карбанионы из ряда перфторолефинов, в том числе из тех, из которых не удавалось получить карбанионы прежде, используя CsF и KF - из перфтор(2-метилпент-2-ена) (31), перфтор(4-метилгепт-3-ена) (32) и перфтор(этилциклогекс-1-ена) (33) [54].
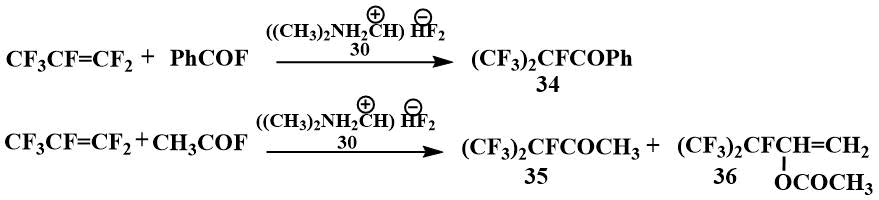
Полученные таким образом перфторалкилкарбанионы были ацилированы фторангидридами карбоновых кислот с получением перфторалкилкетонов и их енольных производных [55].
Можно отметить, что перфторпропилен реагирует с бензоилфторидом в присутствии ТМФБФ уже при комнатной температуре [56], в то время как в присутствии фтористого калия эта реакция осуществляется под давлением при 120°С [57].
В случае ацилирования перфторпропена бензоилфторидом получают перфторизопропилфенилкетон (34), в случае ацилирования ацетилфторидом смесь кетона (35) с ацетатом его енольной формы (36) [55].
Используя ТМФБФ удалось также ацилировать перфтортретбутил анион. При ацилировании перфторизобутилена ацетилфторидом образуются только ацетат (37) и перфторизобутиленолят (38) соответствующего енола. [55]
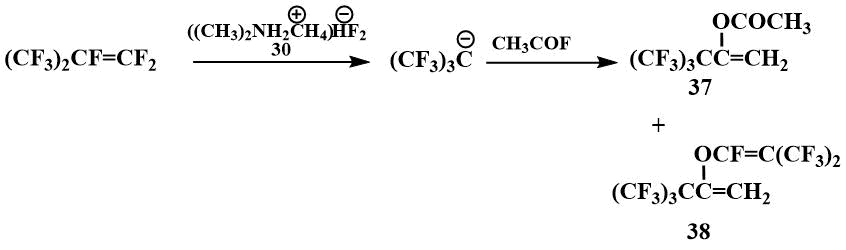
Ацилаты енолов (37) при кипячении с H2SO4 с количественным выходом превращаются в соответствующие кетоны [55].
2.2. Бис-(диметиламино)дифторметан (39) и его циклический аналог 2,2-дифторо-1,3-диметил имидазолин (40).
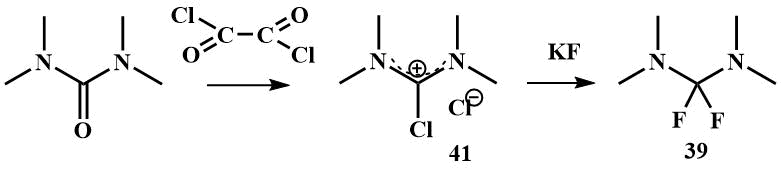
Бис(диметиламино)дифторметан (39) может быть получен фторированием KF тетраметилхлорформамидиний хлорида (41), легко доступного хлорированием тетраметилмочевины каким-либо хлорирующим агентом, подобным образом получают и его циклический аналог (40) [58, 59].
2,2-Дифторо-1,3-диметилимидазолин (40) был применен в качестве мягкого фторирующего агента, он реагирует со алкилкарбинолами подобно реактиву Яровенко и остальным ФАР, а также с карбонильными соединениями. Из бензальдегида с хорошим выходом получен дифторметилбензол, в реакциях же с карбонильными соединениями, содержащими атомы водорода у соседнего атома углерода, наблюдаются побочные реакции, приводящие к образованию алкилвинилфторидов [59].
Взаимодействием бис-(диметиламино)дифторметана (39) с трифторидом фосфора Рошенталлер получил гексакоординированный карбеновый комплекс P(V)-фторида (42) и предложил механизм его образования, в котором на первом этапе фтор-ион переносится из амина (39) к трифториду фосфора с образованием фосфоранид-иона и катиона 2‑фторамидиния, далее катион атакуется фосфоранид-ионом и образуется связь C-P, после чего фосфор отнимает еще один фтор-ион и образут комплекс фосфора (V) с карбеном из амина (39). Подтверждением предложенного механизма явилась фиксация с помощью низкотемпературного ЯМР тетрафторфосфоранид-аниона в растворе [60].Такой же комплекс получен и из 2,2-дифторо-1,3-диметил имидазолина (40) [60].
Полученные гексакоординированные комплексы фосфора представляют собой бесцветные твердые вещества, устойчивые и слабо растворимые в воде.
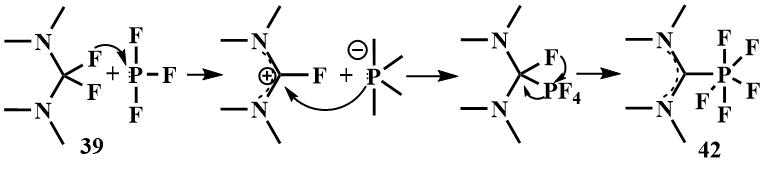
Теми же авторами были получены аналогичные комплексы с элементами главной подгруппы IV группы – германием и оловом – соединения (43) и (44) окислительным присоединением бис-(диалкиламино)дифторметанов к соответствующим галогенидам [58]
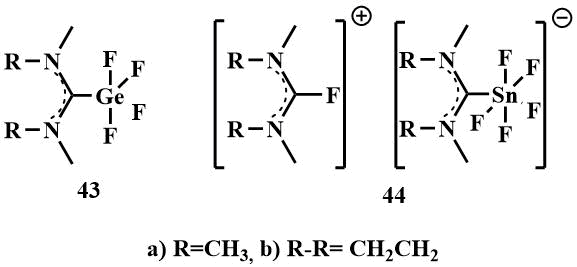
При этом если с германием выделяют нерастворимые в ТГФ твердые комплексы (43 a и b), соответствующие нейтральные комплексы олова не получены. Взаимодействие (39) и (40) с SnF2 приводит к раствору соли (44), связано это вероятно с тем, что тетрафторид олова (IV) является более сильной кислотой Льюиса и способен оторвать дополнительный F -ион из бис-диметиламинодифторметана (39) или 2,2-дифторо-1,3-диметил имидазолина (40) с образованием гигроскопичных солей (44 a и b) с 2- фторамидинием, выступающим в качестве противоиона.
2.3.Трис-диметиламинофторметан (гуанидиний фторид) (46)
Трис-(диметиламино)фторметан (46) представляет особый интерес для генерации фтор-ионов. Впервые он был получен Игумновым реакцией соли пивалата гексаметилгуанидиния (45) с диметиламинотрифторсульфураном и выделен в виде гигроксопичных кристаллов [61].
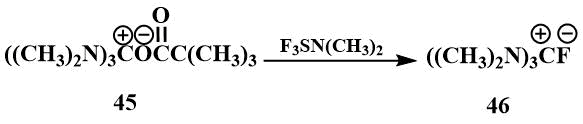
Физические свойства (46) оказались близки к свойствам известных солей гуанидиния. Полученная соль хорошо растворялась в полярных растворителях, таких как ДМФА и ацетонитрил, но не растворялась в эфире. В ИК спектре соединения наблюдали полосу, соответствующую С=N связи в солях гуанидиния. В ПМР спектре имелся только один сигнал, соответствующий CH3N в солях гуанидиния, а в 19F спектре два сигнала - синглет - 25 м.д. (от CF3COOH), соответствующий не связанному прочной водородной связью фтор-иону. и дублет. в области 71 м.д. (от CF3COOH), что дало основание предполагать, что эта соль существует частично в виде гидрофторида, такие сдвиги и константа характерны для HF2 [62].
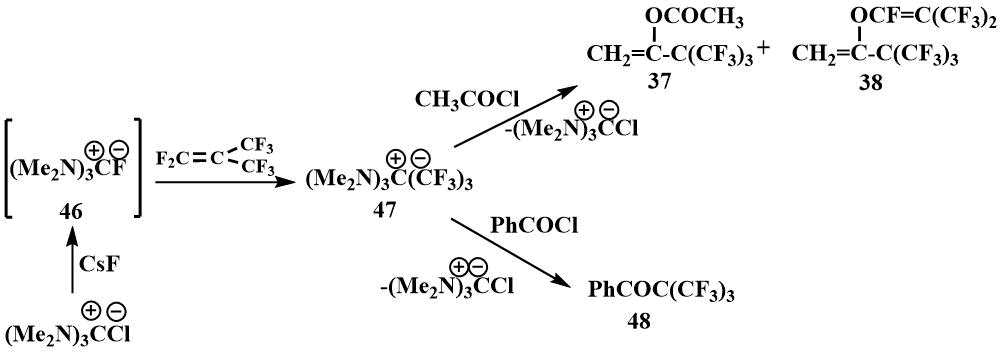
Подобно тетраметилформамидиний бифториду гексаметилгуанидиний фторид реагирует с перфторизобутиленом с образованием ионного соединения, где карбанион стабилизирован гексаметилтриаминометановым катионом - (47) [61].
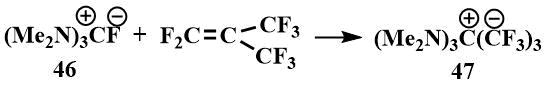
Это же соединение (47) было получено взаимодействием гуанидиний хлорида с цезием фтористым и перфторизобутиленом в диглиме. Образование соли (47) в этом случае также подтверждено ацилированием ее хлористым ацетилом с образованием енольных производных (37) и (38) и хлористым бензоилом, в результате чего был получен кетон (48) [61]. Получение фенилперфтортретбутилкетона (48), который как было показано распадается на перфторизобутилен и фтористый бензоил в присутствии следов фтор-иона [63], свидетельствует о необратимости присоединения перфторизобутилена к гексаметилгуанидинийфториду [61].
Также полученный in situ взаимодействием гуанидиний хлорида с цезием фтористым или калием фтористым в диметилформамиде гуанидиний фторид может быть использован в качестве катализатора фторирования.
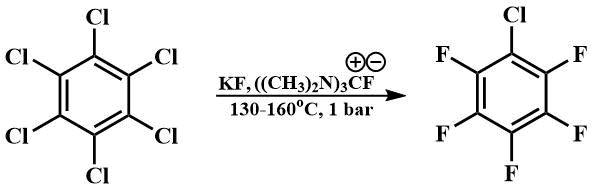
Так с использованием его в качестве катализатора фторирования, была разработан технология фторирования ароматических соединений фтористым калием с непрерывным отбором целевых продуктов, которая позволила снизить температуру фторирования гексахлорбензола с 300°С [64] до 130-160°С и отказаться от использования автоклавов. Эта технология легла в основу производства фторированной ароматики в Пермском филиале РНЦ Прикладная Химия [65].
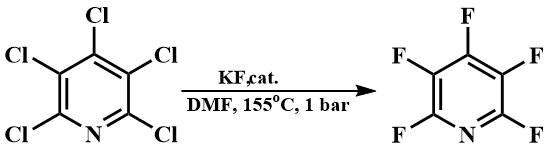
Используя полученный таким образом гексаметилгуанидиний фторид или подобный гексаэтилгуанидиний фторид, удается также провести фторирование пентахлорпиридина в ДМФА при температуре его кипения [66], в то время как без катализатора требуется температура 200°С. [67].
Трис-(диметиламино)фторметан впоследствии привлек внимание многих исследователей.
Чанг с коллегами попыталиссь получить его взаимодействием гуанидиний гидроксида, полученного взаимодействием гуанидиний хлорида с гидроксидом серебра, с водным раствором фтористого водорода в стекле, что ожидаемо привело к гексафторсиликату гуанидиния, который они и выделили в виде его гексагидрата и изучили рентгенографически [68].
В 2000 г. Рёшенталлер сообщил о получении и выделении в кристаллическом виде гексаметилгуанидиний фторида взаимодействием бис-(диметиламино)дифторметана (39) с диметиламинотриметилсиланом [69], однако позже они установили, что выделенная соль представляла собой дифтортриметилсиликат (49), т.е. фактически комплекс гуанидиний фторида с фтортриметилсиланом [70].
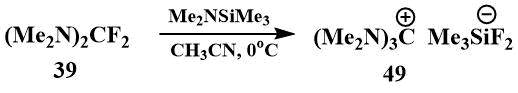
Полученный ими гексаметилгуанидиний дифтортриметилсиликат (фактически комплекс гуанидиний фторида с фтортриметилсиланом) также является источником фтор-иона, реагирует с трифторацетилфторидом и гексафторацетоном в ацетонитриле, давая устойчивые соли. Реакцией соли (Me2N)3C+C2F5O- с метилфтрифлатом при 20С получен с выходом 95% CF3CF2OCH3, с трифторэтилтрифлатом - CF3CF2OCH2CF3. Эти эфиры в последнее время привлекают значительное внимание, поскольку могут заменить запрещенные к использованию хлорсодержащие фреоны [70].
Гуанидиний фторид был также получен взаимодействием борфторида гуанидиния [C(NMe2)3]BF4 с высушенным распылением фтористым калием в растворе абсолютного метанола, но при попытке выделить его, упарив метанол, получили только бифторид [C(NMe2)3]HF2. [70].
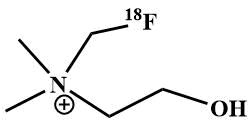
Можно отметить еще одно востребованное соединение, представляющее собой α‑фторамин ‑ [18F] фторметил-диметил-2-гидроксиэтиламмоний – [18F]-фторметилхолин, которое используется в качестве радиофармпрепарата для визуализирования метаболизма клеточной мембраны при диагностике рака простаты и мозга методом ПЭТ [71].
Таким образом необходимо отметить, что α-фторалкиламины, как ковалентные, так и ионные, существенно расширили выбор источников фтор-иона. Они продолжают находить широкое синтетическое применение в реакциях нуклеофильного фторирования и перфторалкоксилирования, предоставляя больше возможностей для управления ходом таких реакций, в частности конденсаций с участием фтор-иона и таким образом повышая синтетические возможности химии фтора.
Благодарности
Работа выполнена при поддержке Министерства науки и высшего образования Российской Федерации.
Литература
- У. Шеппард, К. Шартс, Органическая химия фтора, "Мир", Москва, 1972, 480 с.
- Puar M.S., J.Chem. Educ., 1970, 47(6), 473-474.
- Parker A.J. Q. Rev. Chem. Soc., 1962,16, 163-187.
- Redwood M.E., Wills S J., Can. J. Chem., 1965, 43(7), 1893-1898.
- Redwood M.E.,Wills S J., Can J Chem., 1967, 45(4), 389-395
- Liotta C.L., JACS, 1974, 96(7), 2250-2252.
- Li, Hui-Yin; Sun, Haoran; DiMagno, Stephen G. e-EROS Encyclopedia of Reagents for Organic Synthesis, 2007, 1-9.
- Böhme H., M.Hilp, Chem. Ber., 1970, 103, 104-111.
- Faucett F.S., Tullock C.W., Coffman D.D., JACS, 1962, 84(22), 4275-4285.
- Arnold Z., Collect. Czech. Chem. Commun., 1963, 28, 2047-2051.
- Harder J.R., Smith W.C., JACS, 1961, 83(16), 3422-3224.
- Tyrra W., J. Fluorine Chemistry, 2001, 109(2), 189-194.
- Pawelke G. J. Fluorine Chemistry, 1991, 52(2), 229-234.
- Dmowski W., Kaminski M., J. Fluorine Chemistry, 1983, 23(3), 207-218.
- Яровенко Н. Н., Ракша М. А., ЖОХ, 1959, 29, 2159–2163. (in Russian)
- Takaoka A., Iwakiri H., Ishikawa N., Bull. Chem. Soc. Jpn., 1979, 52, 3377–3380.
- Petrov V. A., Adv. Org. Synth., 2006, 2, 269–290.
- Haszeldine R. N., J.Chem.Soc., 1951, 102-104.
- Livingston, R. L.; Vaughan, G., JACS, 1956, 78, 4866-9.
- Bürger, H.; Eujen, R.; Niepel, H.; Pawelke, G., J. Fluorine Chemistry, 1981, 17(1), 65-74.
- Satori P., Velayutham D., Ignat'ev N.; Noel M., J. Fluorine Chemistry, 1997, 83(1), 1-8.
- Молдавский Д.Д., Фурин Г.Г., Шкультетская Л.В., Эйфман Б.Я., ЖПХ, 2002, 75(6), 959‑961.
- Ягупольский Л.М., Кондратенко Н.В., Дронкина М.И., Ягупольский Ю.Л., ЖОрХ, 1980, 16(12), 2508.
- Brauer D. J., Bürger M, . Grunwald M., Pawelke G., J. Wilke J., Z. Anorg. Allg. Chem. 1986, 537, 63-78.
- Oberhammer H., Mendeleev Commun., 2006, 16(3), 136–137.
- А.В. Фокин, Ю.Н. Студнев, А.И. Рапкин, Д.А.Султанбеков, Т.М. Потарина, Изв. АН СССР, Сер. хим., 1984, 2, 411-415.
- Фокин А.В., Зимин В.И., Студнев Ю.Н., Султанбеков Д.А., ЖОХ, 1968, 38(7), 1510-1511.
- V. A. Petrov, S. Swearingen, W. Hong and W. Chris Petersen, J. Fluorine Chem., 2001, 109, 25–31.
- Schmitt E., Panossian A., Vors J.-P., Funke C., Lui N., Pazenok S., Leroux F.R., Chem. A Eur. J., 2016, 22, 11239-11244.
- Takaoka A., Iwamoto K., Kitazume T., Ishikawa N., J. Fluorine Chem., 1979,14, 421-428.
- Aribi F., Schmitt E., Panossian A., Vors J.-P., Pazenok S., Leroux F.R., Org. Chem. Front., 2016, 3, 1392–1415.
- Liska F., Chemicke Listy, 1972, 66(2), 189-197.
- Dax, K. Science of Synthesis, 2006, 2005, 34, 71-148.
- Igumnov S., Kornilov V., Fluorine Notes, 2000, 1(8).
- Caster K. C., Zefirov N. S., Lermontov S. A., Filler R., N,N‐Diethyl‐2‐chloro‐1,1,2‐trifluoroethylamine, e-EROS Encyclopedia of Reagents for Organic Synthesis, 2009, 1-3 https://doi.org/10.1002/047084289X.rd184.
- Commare B., Schmitt E., Aribi F., Panossian A., Pazenok S., Leroux F.R., Molecules, 2017, 22, 977-1003.
- Filler R., Hofferberth J., Beckett J., N,N‐Diethyl‐1,1,2,3,3,3‐hexafluoropropylamine, e-EROS Encyclopedia of Reagents for Organic Synthesis, 2007, https://doi.org/10.1002/9780470842898.rd196.pub2.
- Junk C.P., Petrov V. A., 1,1,2,2‐Tetrafluoroethyl‐N,N‐dimethylamine, e-EROS Encyclopedia of Reagents for Organic Synthesis, 2014, https://doi.org/10.1002/047084289X.rn01690.
- Böhme H., Hartke K., Chem. Ber., 1960, 93, 1305-1309.
- Кнунянц И. Л., Делягина Н. И., Игумнов С. М., Изв. AH СССР. Сер. хим., 1981, 4, 857‑859.
- Игумнов, С.М, Делягина Н.И., Зейфман Ю.В. , Кнунянц И.Л. , Изв. АН СССР, Сер. хим.,1984, 4, 827-832.
- Игумнов С.М., Леконцева Г.И., Шипигусев А.А., Мухаметшин В.Ф., ЖПХ, 2005, 78(3), 438-440.
- Igoumnov S.M., Fluorine Notes, 2006, 46, 3-4.
- Синтезы фторорганических соединений, под ред. И.Л. Кнунянца и Г.Г. Якобсона. М; Химия, 1973, 312 с.
- Allefeld N., Neumann B., Stammler H.-G., Röschenthaler G.-V., Ignat’ev N., and Hoge B., Chem. A Eur. J. 2014, 20, 7736-7745.
- Aravindan V., Gnanaraj J., Madhavi S., and Liu H.-K., Chem. Eur. J. 2011, 17, 14326-14346.
- Arnold Z., Coll.Czech. Chem. Comm, 1959, 24, 4048-4049.
- Кнунянц И.Л., Делягина Н.И., Игумнов С.М., Изв. АН СССР., Сер. хим., 1981, 4, 860‑863.
- Патент CN111153867A, 2020.
- Патент CN111635321, 2020.
- Патент US2957001, 1957.
- Ягупольский Л.М., Кондратенко Н.В., Тимофеева Г.Н., Дронкина М.И., Ягупольский Ю.Л., ЖОрХ, 1980, 16(12), 2508-2513.
- Dmowski W., Kaminski M., J Fluorine Chem. 23, 1983, 219-228.
- Делягина Н.И., Игумнов С.М., Снегирев В.Ф., Кнунянц И.Л., Изв. АН СССР, Сер. хим., 1981, 10, 2238-2243.
- Игумнов С.М. , Делягина Н.И., Кнунянц И.Л., Изв. АН СССР, Сер. хим., 1981, 10, 2339‑2342.
- Игумнов С.М. Новые источники фторид-иона, стабилизированные органическими катионами: диссертация кандидата хим. наук, Москва, 1983, 145 с.
- Ishikawa N., Shin S., Bull.Chem. Soc.Jpn, 1975, 48(4), 1339-1340.
- Böttcher T., Bassil B.S., Zhechkov, Röschenthaler G.-V., Inorganic Chemistry, 2012, 51(2), 763-765.
- Hidetoshi Hayashi, Hiroshi Sonoda, Kouki Fukumura and Teruyuki Nagata, Chem. Comn., 2002, 1619-1619.
- Böttcher T., Shyshkov O., Bremer M., Bassil B. S., Röschenthaler G.-V., Organometallics, 2012, 31(4), 1278-1280.
- Игумнов С.М. , Делягина Н.И., Кнунянц И.Л., Изв. АН СССР, Сер. хим., 1986, 6,, 1315-1317.
- Fujiwara F.Y., Vartin J.S., JACS, 1974, 96(25), 7625-7626.
- Кнунянц И.Л., Зейфман Ю.В., Ланцева Л. Г., Докл. АН СССР., 1980, 254(1), 117-120.
- Ворожцов Н.Н.-мл., Платонов В. Е., Якобсон Г. Г., Изв. AH СССР., 1963, 8, 1524.
- Игумнов С.M., Заболотских А.В., Патент РФ 2164508 (2001).
- Синтезы фторорганических соединений, ч.4, под ред. Игумнова С.М., Игумновой Э.В., Москва, ЗАО ПиМ Инвест, 2018, 222 с.
- Chambers K. D., Hutchinson J., and Musgrave W. K. R., Chem Soc 1964, 3573.
- Zhang, R. Bau,J.A,Sheehy, K.O.Christe J., Fluor.Chem., 1999, 98, 121-126.
- Kolomeitsev A.A., Bissky G., Kirsch P., Röschenthaler G.-V., Journal of Fluorine Chemistry, 2000, 103, 159-161.
- Kolomeitsev A. A., Bissky G., Barten J., Kalinovich N, Lork E., Röschenthaler G.-V., Inorganic Chemistry, 2002, 41(23), 6118-6124.
- Fedorova O. S., Vaitekhovich F. P., Krasikova R. N., Pharmaceutical Chemistry Journal, 2018, 52(8), 730-734.
Статья рекомендована к публикации членом редколлегии к.х.н. М. А. Манаенковой
Fluorine Notes, 2021, 139, 3-4


