Поступило в редакцию: Декабрь 2020
УДК 17677/fn20714807.2021.01.03
Fluorine Notes, 2021, 134, 5-6
УСОВЕРШЕНСТВОВАННЫЙ СИНТЕЗ 3,3- И 5,5-ДИФТОР-2-АМИНОЦИКЛОГЕКСАНКАРБОКСИЛАТОВ И РАСШИРЕНИЕ ВОЗМОЖНОСТЕЙ ЭТОГО МЕТОДА С ПОМОЩЬЮ СЕЛЕНООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Attila M. Remete,a,b Melinda Nonn,a,b,c Anas Semghouli,a Reijo Sillanpää,d Loránd Kissa,b*
aInstitute of Pharmaceutical Chemistry, University of Szeged, H-6720 Szeged, Eötvös u. 6, Hungary
kiss.lorand@szte.hu
bInterdisciplinary Excellence Centre, Institute of Pharmaceutical Chemistry, University of Szeged, H-6720 Szeged, Eötvös u. 6, Hungary
cMTA-SZTE Stereochemistry Research Group, Hungarian Academy of Sciences, H-6720 Szeged, Eötvös u. 6, Hungary
dDepartment of Chemistry, University of Jyväskylä, FIN-40014, Jyväskylä, Finland
Графическая аннотация:
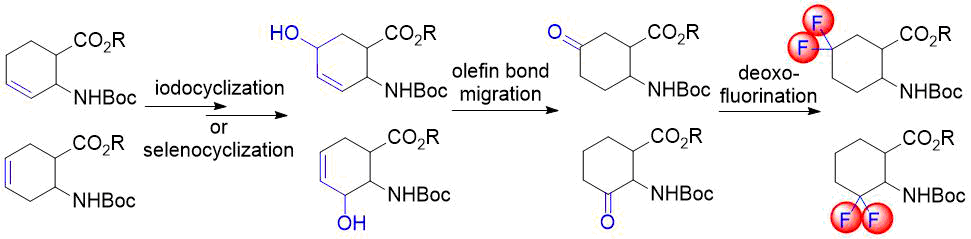
Аннотация: Путь синтеза к гем-дифторированным циклогексановым β-аминокислотам, состоящий из процессов регио- и стереоселективного гидроксилирования, восстановления связей C = C, окисления спирта и дезоксофторирования, описанные ранее, был значительно усовершенствован. За счет миграции двойной связи, катализируемой Pd, промежуточные аллиловые спирты непосредственно и эффективно преобразуются в соответствующие насыщенные кетоны. Для расширения возможностей предложенного метода была предпринята попытка синтеза новых производных циклических β-аминокислот с фрагментом аллилового спирта путем использования селенорганических соединений.
Ключевые слова: β-аминокислоты, деоксофторирование, миграция двойной связи, окислительное деселенилирование, селеноциклизация.
Введение
Несмотря на то, что фторсодержащие органические соединения в природе встречаются довольно редко [1], их все чаще стали использовать в фармацевтической химии [2-5]. Причины успеха фторированных лекарственных средств - это преимущества фторирования, которые обусловлены уникальными свойствами атома фтора и связи типа «углерод-фтор» прежде всего из-за небольшого размера атома фтора, который способен изостерически заменять атомы водорода, гидрокси- или метокси- группы. Важно отметить, что введение фтора может эффективно подавлять процесс окислительного метаболизма, поскольку связи C – F прочнее связей C – H, а сверхвысокая электроотрицательность фтора снижает электронную плотность окружающих атомов. Введение фтора также делает более кислыми близлежащие функциональные группы, изменяя соотношение между заряженными и нейтральными лекарственными компонентами. Вместе с полезным полярным гидрофобным характером мотива C – F это изменение влияет на липофильность. Атом фтора в связях C – F с частичным отрицательным зарядом также может создавать новые полезные перспективные электростатические взаимодействия с целевыми молекулами [2, 5, 6].
Аминокислоты имеют большое значение для медицинской химии [7-20], в весьма разнообразном семействе соединений которых в последние десятилетия [9‑13,20] все большее внимание к себе стали привлекать циклические β-аминокислоты.
Эти соединения в виде субъединиц могут содержаться в биологически активных природных соединениях, например, в бластицидине S (1), амипуримицине (2), гугеротине и хрискандине (см. Схему 1) [9,10,21]. Многие низкомолекулярные производные β-аминокислот, включая такие натуральные соединения, как циспентацин (3) и оксетин, а также такие синтетические соединения, как икофунгипен и тилидин (4), также обладают биологической активностью (см. Схему 1) [9,10]. К весьма перспективным применениям циклических β-аминокислот можно отнести формирование β-пептидов, обладающих высокой устойчивостью к ферментативному расщеплению, и фолдамеров [11-13].
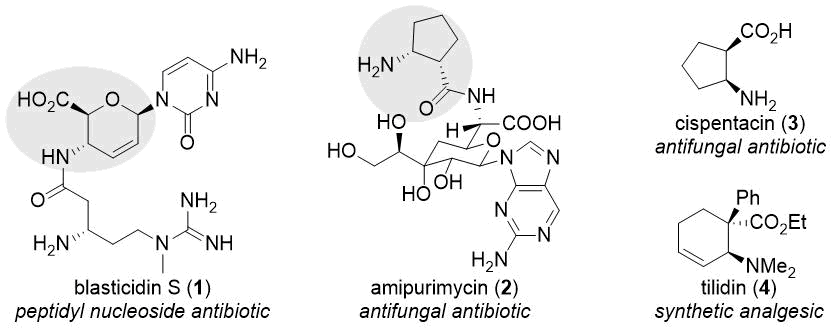
Схема 1. Биоактивные производные β-аминокислот.
Поскольку как фторорганические соединения, так и аминокислоты обладают большой ценностью, производным фторированных аминокислот в настоящее время уделяется все большее внимание. Наиболее изученные семейства соединений - это фторированные ациклические или циклические α-аминокислоты и фторированные ациклические β-аминокислоты [14-19]. Поскольку фторированные циклические β-аминокислоты практически не изучены [19-20], авторы в последнее десятилетие свое внимание сосредоточили на синтезе именно этих соединений и разработали значительное количество методов синтеза фторированных производных циклических β-аминокислот [22-37].
Обсуждение результатов
В более ранних своих работах авторы сообщали о синтезе гем-дифторированных производных β-аминокислоты циклогексана из β-аминокислот (±)-5 и (±)-14 циклогексена посредством регио- и стереоселективного гидроксилирования, восстановления связи C = C, окисления спирта и дезоксофторирования (см. Схемы 2 и 3) [22,23,38].
Синтез гем-дифторированной производной β-аминокислоты циклопентана (±)-37 из циклопентеновых β-аминокислот (±)-26 и (±)-27 был осуществлен по несколько путем (см. Схему 4) [26].
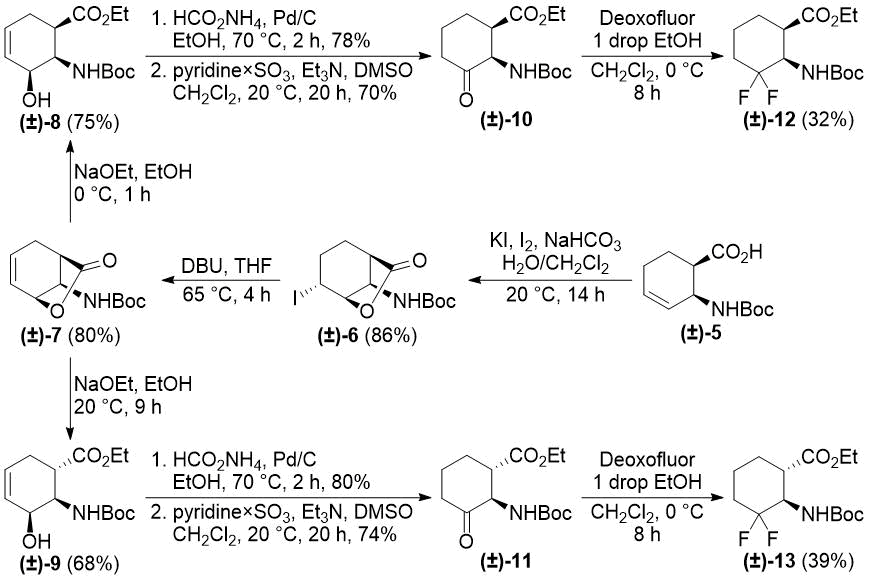
Схема 2. Исходный синтез 3,3-дифторированных β-аминоэфиров циклогексана(см. [22]).
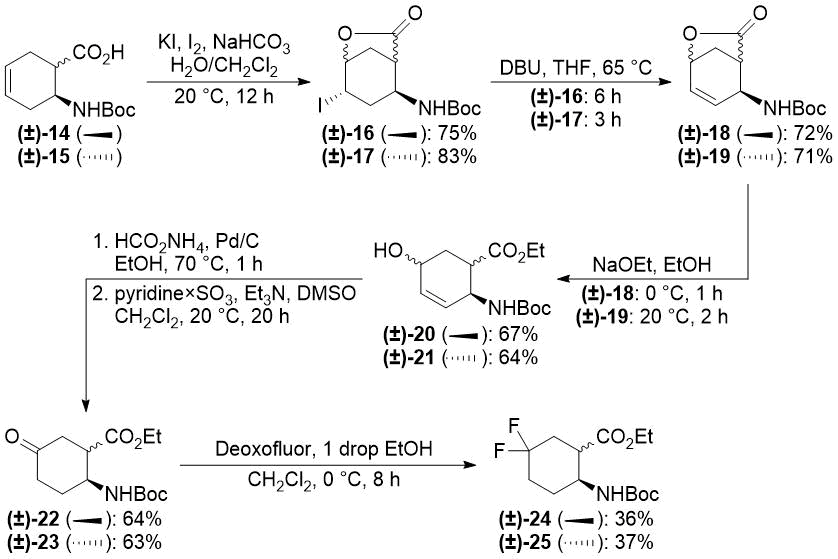
Схема 3. Исходный синтез 5,5-дифторированных β-аминоэфиров циклогексана (см. [23]).
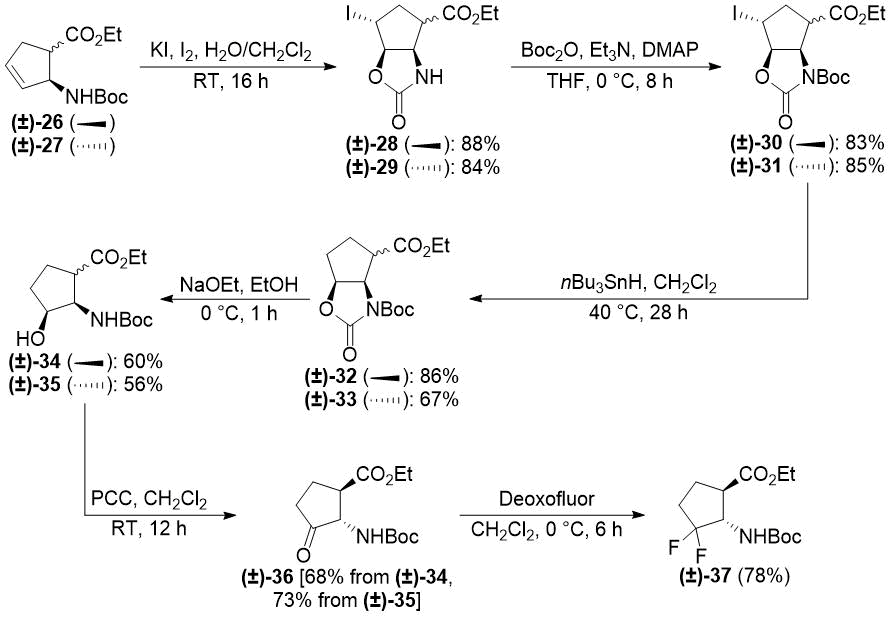
Схема 4. Исходный синтез гем-дифторированного β-аминоэфира циклопентана(±)-37 (см. [26]).
Однако мы поняли, что у этих методов есть некоторые проблемы. В способах, изображенных на Схемах 2 и 3, окислительно-восстановительно-нейтральные процессы (превращения аллильных спиртов (±)-8, (±)-9, (±)-20 и (±)-21 в кетоны (±)-10, (±)-11, (±)-22 и (±)-23) выполнялись в два последовательных этапа. Этот подход не является полностью оптимальным с экономической точки зрения, поскольку в синтезе, изображенном на Схеме 4, используются стехиометрические количества токсичного гидрида трибутилолова и опасного хлорхромата пиридиния (PCC).
Цель проведения исследований состояла в максимальном устранении указанных недостатков. Так, внимание было привлечено к катализируемому переходными металлами прямому превращению аллиловых спиртов в кетоны. Этот простой атом-экономичный процесс основан на изомеризации связи C = C в кольце субстрата с получением промежуточного енола, который таутомеризуется в более стабильную оксоформу (см. Рисунок 1). Непосредственной причиной этого превращения является более высокая прочность связи C = O по сравнению с прочностью связи C = C. Подобные реакции в основном протекают при гомогенном катализе, однако, как сообщалось, оказалось успешным и также использование таких гетерогенных катализаторов, таких как Pd/C [39], Pd/TiO2 [40], Pd/Al2O3 [41–42] или наночастицы Pd [43]. Дополнительным преимуществом этого превращения является простота обращения и разделения гетерогенных катализаторов, поэтому было принято решение об апробации окислительно-восстановительной изомеризации аллиловых спиртов с использованием катализатора Pd/C. Возможными побочными реакциями при этом являются реакции восстановления связи C = C субстрата или связи C = O продукта, поэтому для максимального увеличения выхода желаемого продукта требовался тщательный выбор условий реакции.
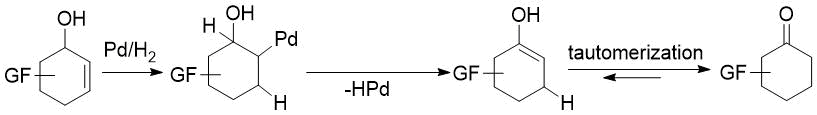
Рисунок 1. Прямое превращение аллиловых спиртов в кетоны за счет миграции связи C = C.
Первым модельным субстратом было производное аллилового спирта (±)-8. Нескольких попыток изменения ряда условий реакции (например, замены растворителя с EtOH на EtOAc и увеличения времени реакции) стало достаточно для сдвига в сторону окислительно-восстановительной изомеризации, после чего продукт (±)-10 был выделен с выходом 89% (см. Схему 5). Отметим, что первоначальное двухэтапное преобразование типа (±)-8 → (±)-10 обеспечивало общий выход продукта только 55%. Аналогичное преобразование соединения (±)-9 в кетон (±)-11 также оказалось успешным (см. Схему 5). Фактический выход в 92% выгодно отличается от 59% выхода при двухэтапном процессе. После этого были преобразованы кетоэфиры (±)-10 и (±)-11. Обработка трифторидом бис(2-метоксиэтил)аминосеры (Deoxofluor) в CH2Cl2 в соответствии с немного измененной методикой в течение 12 ч при комнатной температуре приводит к получению соответствующих дифторированных производных (±)-12 и (±)-13 (См. Схему 5).
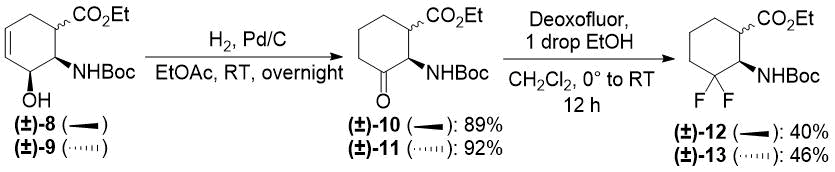
Схема 5. Одностадийная окислительно-восстановительная изомеризация аллиловых спиртов (±)-8 и (±)-9 до соответствующих кетопроизводных.
Процесс окислительно-восстановительной изомеризации аллиловых спиртов (±)-20 и (±)-21 также протекал успешно (см. Схему 6), обеспечивая гораздо более высокие выходы, чем при использовании исходного протокола [(±)-20 → (±)-22: 64% (двустадийный метод) против 91% (новый одностадийный метод); (±)-21 → (±)-23: 63% (двустадийный метод) против 90% (новый одностадийный метод)]. Оба оксоэфира (±)-22 и (±)-23 в результате дезоксофторирования легко превращались в желаемые дифторированные продукты (±)-24 и (±)-25 (см. Схему 6).
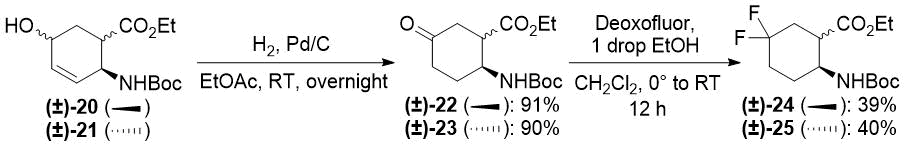
Схема 6. Путь одноэтапной реакции окислительно-восстановительной изомеризации аллиловых спиртов (±)-20 и (±)-21 до соответствующих кетопроизводных
Для расширения возможностей этого нового метода авторы пытались синтезировать другие циклические β-аминоэфиры с фрагментами аллилового спирта. К сожалению, подвергнуть оксазолидинон (±)-30 элиминированию HI / открытию гетероцикла (см. Схемы 2 и 3) не удалось, поскольку ни t-BuOK, ни DBU не были способны индуцировать отщеплние HI. Обработка (±)-30 с помощью КОН в THF/H2O дала неожиданный продукт (±)-39 через раскрытие карбаматного цикла и SN2 замещения йода (см. Схему 7). Отметим, что в более раннем исследовании авторов диол (±)-39 не мог быть получен путем син-дигидроксилирования аминоэфира (±)-26 с использованием OsO4/NMO [44].
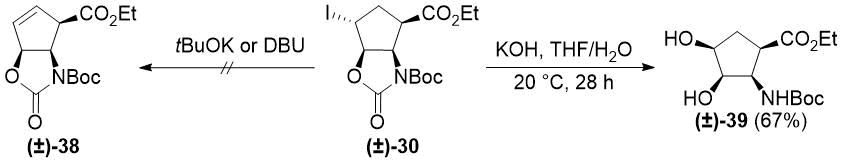
Схема 7. Реакции йодоксазолидинона (±)-30 с основаниями.
Для решения проблем элиминирования (отщепления) авторы обратились к опыту селенорганической химии. В аналогичных электрофильных процессах PhSe+ может принимать участие как I+ и удаляться при мягких условиях окисления (например, с помощью H2O2) [45–47]. Результат обработки эфиров β-аминоциклопентена с помощью PhSeBr зависел от защитной группы. Из N-бензоилированного продукта (±)-40 образуется производное селеноксазолина (±)-41, в то время как карбаматные защитные группы (в особенности Boc) приводят к образованию селеноксазолидинона (±)-44. N-Boc защита соединения (±)-44 с его последующим окислительным деселенилированием в основных условиях [47] давала требуемый продукт (±)-38 (см. Схему 8).
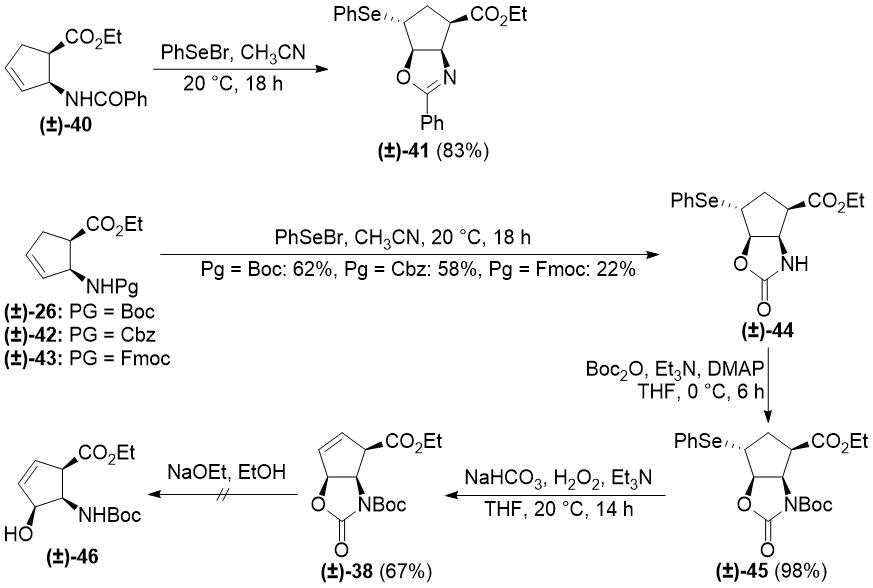
Схема 8. Путь селеноциклизации цис-циклопентеновых β-аминоэфиров.
К сожалению, все попытки открытия гетероцикла (±)-38 с помощью этоксида приводили к его разложению с образованием неидентифицируемых полимерных материалов.
Аналогичные преобразования N-Boc-защищенного трансаминоэфира (±)-27 приводили к образованию ненасыщенного оксазолидинона (±)-49 (см. Схему 9). Помимо ЯМР-анализа, структура промежуточного соединения (±)-48 была подтверждена рентгеноструктурным анализом его кристаллической структуры (см. Рисунок 2). Подробности определения его структуры представлены в экспериментальной части данной работы. Подобно его стереоизомеру (±)-38, обработка (±)-49 с помощью этоксида вместо разложения приводила к раскрытию гетероцикла.
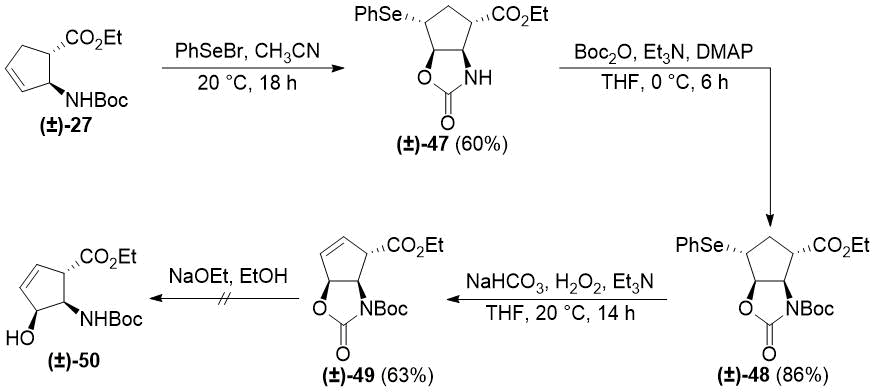
Схема 9. Селеноциклизация транс-циклопентенового β-аминоэфира (±)-27.
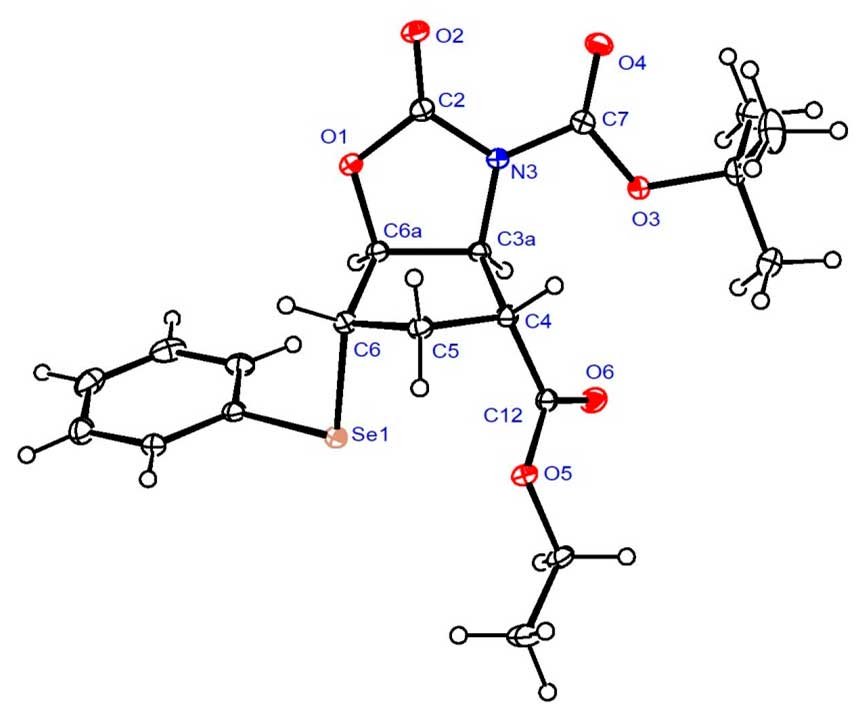
Рисунок 2. Кристаллическая структура селенооксазолидинона (±)-48.
После этого к эфиру циклогексен-β-амину (±)-51 были последовательно применены операции селеноциклизации/N-защиты/окислительного деселенилирования. При этом окисление неожиданно привело к образованию сверхненасыщенного эфира (±)-55, наиболее правдоподобным объяснением чего является E1cb элиминирование в требуемом продукте (±)-54 под воздействием основания (см. Схему 10).
Разложение (±)-38 и (±)-49 при их обработке высокощелочным этоксидом можно объяснить аналогичными процессами E1cb элиминирования (получаемые крайне реакционно-способные α, β, γ, δ-ненасыщенные сложные эфиры легко подвергаются полимеризации).
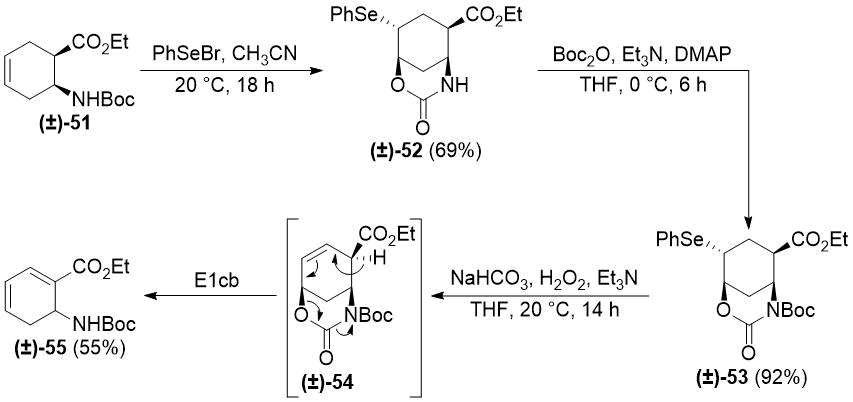
Схема 10. Селеноциклизация β-аминоэфира циклогексена (±)-51.
Во избежание E1cb элиминирования перед окислительным деселенилированием можно выполнять раскрытие гетероцикла и заменять плохо уходящую гидроксильную группу на приемлемо уходящий карбамат. Даже в этом случае может оказаться необходимым провести окисление в кислых условиях (например, в среде MCPBA или H2O2/CF3COOH [46]) вместо основных условий, применявшихся выше. Эти исследования в настоящее время продолжаются в нашей лаборатории.
Выводы
Используя процесс одноэтапной окислительно-восстановительной изомеризации аллиловых спиртов в насыщенные кетоны, мы значительно улучшили синтез цис- и транс-изомеров 3,3- и 5,5-дифторированных 2-аминоциклогексанкарбоксилатов. Для расширения возможностей этого усовершенствованного метода были предприняты попытки получения различных β-аминоциклоалканкарбоксилатов с фрагментом аллилового спирта с использованием методов селенорганической химии. Некоторые ключевые промежуточные соединения имели тенденцию к E1cb элиминированию, и желаемые продукты не образовывались. Изменение синтетического пути синтеза с целью устранения источника проблемы все еще продолжается. В настоящее время исследуются другие варианты методов синтеза, связанные с развитием методов доступа к гем-дифторированным структурам.
Экспериментальная часть
Общая информация
Химические реактивы были куплены у компании Sigma-Aldrich. Растворители использовались в том же виде, что и при поставке. Температуры плавления определялись на аппаратуре Кофлера. Элементный анализ выполняют на анализаторе компании Perkin – Elmer (модель CHNS-2400 Ser II). Силикагель 60 F254 приобретался у компании Merck. Спектры ЯМР регистрировались при комнатной температуре на спектрометре компании Bruker (модель Avance 400) на частоте 400,13 МГц для 1H и на частоте 100,76 МГц для 13C, в растворе CDCl3 или D6-DMSO, с использованием для фиксации поля сигнала от дейтерия в растворителе. Химические сдвиги для 1H и 13C приводятся относительно ТМС. Производные β-аминокислот (±)-5-(±)-37, (±)-40, (±)-42, (±)-43 и (±)-51 - известные соединения, ранее описанные в литературе [22,23,26,38,48-50].
Общая методика изомеризации, катализируемой Pd
В раствор 1 ммоль аллилового спирта в 10 мл EtOAc вводят 15 мг Pd/C (10 % по массе). Реакционную смесь перемешивают при температуре 20°C в атмосфере водорода (при давлении 1 бар) в течение ночи, а затем фильтруют через стеклянный фильтр, покрытый Celite®. Полученный фильтрат сушат над Na2SO4 и концентрируют. Неочищенный продукт очищают методом колоночной хроматографии на силикагеле (н-гексан/EtOAc).
Общая методика селеноциклизации
К раствору 5 ммоль β-аминоэфира в 25 мл MeCN добавляют 5 ммоль PhSeBr, а полученную смесь перемешивают при температуре 20°C в течение 18 ч. За ходом реакции наблюдают с помощью метода тонкослойной хроматографии. После завершения перемешивания реакционную смесь разбавляют 60 мл EtOAc и промывают 2×20 мл насыщенного водного раствора NaHCO3. Органическую фазу сушат над Na2SO4 и концентрируют. Неочищенный продукт кристаллизуют из Et2O.
Общая методика для N-Boc защиты продуктов селеноциклизации
В раствор, состоящий из 30 мл ТГФ и 1,5 г селеноциклизированного соединения. вводят при температуре 0°C 2 экв. Et3N, 1 экв. Boc2O и 20 мол.% DMAP. Полученной смеси дают возможность нагреться до 20°C, затем перемешивают в течение 6 ч, реакционную смесь разбавляют 70 мл EtOAc и промывают водой 3×30 мл. Органическую фазу сушат над Na2SO4 и концентрируют. Неочищенный продукт очищают методом колоночной хроматографии на силикагеле (н-гексан/EtOAc).
Общая методика окислительного деселенилирования
В раствор 2 ммоль фенилселенилового β-аминоэфира в 25 мл ТГФ вводят 2 экв. NaHCO3 и 5 экв. H2O2 (в виде 30%-ного водного раствора) при температуре 0°C. Смеси дают возможность нагреться до 20°C, затем перемешивают в течение 30 мин. После добавления 2,5 экв. Et3N перемешивание продолжают еще в течение 14 ч, реакционную смесь разбавляют 90 мл EtOAc и промывают 2×20 мл воды. Органическую фазу сушат над Na2SO4 и концентрируют. Неочищенный продукт очищают методом колоночной хроматографии на силикагеле (н-гексан/EtOAc).
Реакция йодоксазолидинона (±)-30 с КОН/THF
В раствор 300 мг соединения (±)-30 в 15 мл ТГФ вводят 1 мл воды и 2 экв. КОН, смесь перемешивают при температуре 20°C в течение 28 ч, реакционную смесь разбавляют 20 мл воды и экстрагируют с помощью 3×10 мл CH2Cl2. Органическую фазу сушат над Na2SO4 и концентрируют. Неочищенный продукт очищают методом колоночной хроматографии на силикагеле (н-гексан/EtOAc).
Общая процедура фторирования кетоаминоэфиров
В раствор аминоэфира (±)-10, (±)-11, (±)-22 или (±)-23 (1,5 ммоль) в CH2Cl2 (15 мл) вводят одну каплю EtOH, затем по каплям при температуре 0°C прибавляют трифторид бис(2-метоксиэтил)аминосеры (Deoxofluor) (1,5 экв., 50% в толуоле), перемешивают при комнатной температуре в течение ночи и разбавляют смесь CH2Cl2 (25 мл). Органическую фазу промывают водным раствором NaHCO3, затем сушат над Na2SO4 и концентрируют. Неочищенный продукт очищают методом колоночной хроматографии на силикагеле (н‑гексан-EtOAc 4: 1).
Рентгеноструктурное исследование (±)-48
Кристаллографические данные регистрируют при 123 К с помощью дифрактометра с детектором, имеющим рабочую область ПЗС Nonius-Kappa и использующим монохроматизированный графит Mo-K-излучения с длиной волны = 0,71073 Å (см. [51]). Структуру расшифровывают с помощью прямых методов с использованием программы SHELXS-97 [2], а полноматричную коррекцию выполняют методом наименьших квадратов на F2 с помощью этой же программы. Атомы водорода CH вводят на фиксированные расстояния от их атомов-матриц с фиксированными параметрами смещения. График ORTEP строят с помощью программы ORTEP-3 for Windows [53]. Номер осаждения CCDC 2051318 содержит дополнительные кристаллографические данные, которые используются в данной работе и которые можно получить бесплатно в Кембриджском центре кристаллографических данных по адресу http://www.ccdc.cam.ac.uk/conts/retrieving.html (или в Кембриджском центре кристаллографических данных, 12 Union Road, Кембридж CB2 1EZ , Великобритания; факс: (международный) + 44-1223-336-033; электронная почта: deposit@ccdc.cam.ac.uk).
Результаты кристаллографических исследований (±)-48, C20H25NO6Se, Mr = 454.37, monoclinic, space group P21/c (no. 14), a = 9.1060(1), b = 18.8854(3), c = 11.8729(2) Å, = 90, β = 90.73(1), = 90, V = 2041.62(5) Å3, T = 123 K, Z = 4, μ(Mo-Kα) = 1.478 mm-1. Total 4450 refelections, unique 3973. Refinement of 4450 reflections (257 parameters) with I > 2(I) converged at final R1 = 0.0236 ( R1 all data = 0.0289), wR2 = 0.0553 (wR2 all data = 0.0575), GOF = 1.049.
Дополнительную информацию по ЯМР спектрам соединений можно найти в английской PDF версии.
Благодарности
Авторы выражают свою благодарность Венгерскому исследовательскому фонду (NKFIH № K 119282) за финансовую поддержку, а также за финансовую поддержку проекта GINOP-2.3.2-15-2016-00038, выполняемого в рамках венгерского гранта EFOP-3.6.1-16-2016-00008, финансируемого ЕС. Авторы также благодарят Министерство людских ресурсов Венгрии за грант 20391/2018/FEKUSTRAT).
References
- Carvalho, M. F., Oliveira, R. S., Crit. Rev. Biotechnol., 2017, 37, 880-897.
- Hagmann, W. K. J., Med. Chem., 2008, 51, 4359-4369.
- Wang, J., Sánchez-Roselló, M., Aceña, J. L., del Pozo, C., Sorochinsky, A. E., Fustero, S., Soloshonok, V. A., Liu, H., Chem Rev., 2014, 114, 2432-2506.
- Zhou, Y., Wang, J., Gu, Z., Wang, S., Zhu, W., Aceña, J. L., Soloshonok, V. A., Izawa, K., Liu, H., Chem Rev., 2016, 116, 422-518.
- Han, J., Remete, A. M., Dobson, L. S., Kiss, L., Izawa, K., Moriwaki, H., Soloshonok, V. A., O’Hagan, D. J., Fluorine Chem., 2020, 239, 109639.
- Purser, S., Moore, P. R., Swallow, S., Gouverneur, V., Chem. Soc. Rev., 2008, 37, 320-330.
- Soloshonok, V. A., Izawa, K. (Eds.), Asymmetric Synthesis and Application of α-Amino Acids, ACS Symposium Series #1009, Oxford University Press, 2009.
- Liu, J., Han, J., Izawa, K., Sato, T., White, S., Meanwell, N. A., Soloshonok, V. A., Eur. J. Med. Chem., 2020, 208, 112736.
- Kiss, L., Fülöp, F. Chem Rev. 2014, 114, 1116-1169.
- Kiss, L., Mándity, I. M., Fülöp, F., Amino Acids, 2017, 49, 1441-1455.
- Martinek, T. A., Fülöp, F., Chem. Soc. Rev., 2012, 41, 687-702.
- Gopalan, R. D., Del Borgo, M. P., Mechler, A. I., Perlmutter, P., Aguilar, M.-I., Chem. Biol., 2015, 22, 1417-1423.
- Lee, M.-R., Raman, N., Gellman, S. H., Lynn, D. M., Palecek, S. P., ACS Chem. Biol., 2014, 9, 1613-1621.
- Salwiczek, M., Nyakatura, E. K., Gerling, U. I. M., Ye, S., Koksch, B., Chem. Soc. Rev., 2012, 41, 2135-2171.
- Moschner, J., Stulberg, V., Fernandes, R., Huhmann, S., Leppkes, J., Koksch, B., Chem. Rev., 2019, 119, 10718-10801.
- Qiu, X.-L., Qing, F.-L., Eur. J. Org. Chem., 2011, 3261-3278.
- Mei, H., Han, J., Klika, K. D., Izawa, K., Sato, T., Meanwell, N. A., Soloshonok, V. A., Eur. J. Med. Chem., 2020, 186, 111826.
- March, T. L., Johnston, M. R., Duggan, P. J., Gardiner, J., Chem. Biodivers., 2012, 9, 2410‑2441.
- Mikami, K., Fustero, S., Sánchez-Roselló, M., Aceña, J. L., Soloshonok, V., Sorochinsky, A., Synthesis, 2011, 19, 3045-3079.
- Kiss, L., Fülöp, F., Chem. Rec., 2018, 18, 266-281.
- Wang, S., Zhang, Q., Zhao, Y., Sun, J., Kang, W., Wang, F., Pan, H., Tang, G., Yu, B., Angew. Chem. Int. Ed., 2019, 58, 10558-10562.
- Kiss, L., Forró, E., Fustero, S., Fülöp, F., Org. Biomol. Chem., 2011, 9, 6528-6534.
- Kiss, L., Forró, E., Fustero, S., Fülöp, F., Eur. J. Org. Chem., 2011, 4993-5001.
- Nonn, M., Kiss, L., Hänninen, M. M., Sillanpää, R., Fülöp, F., Chem. Biodivers., 2012, 9, 2571-2580.
- Kiss, L., Nonn, M., Sillanpää, R., Fustero, S., Fülöp, F. Beilstein J., Org. Chem., 2013, 9, 1164‑1169.
- Kiss, L., Nonn, M., Forró, E., Sillanpää, R., Fustero, S., Fülöp, F., Eur. J. Org. Chem., 2014, 19, 4070-4076.
- Nonn, M., Kiss, L., Haukka, M., Fustero, S., Fülöp, F., Org. Lett., 2015, 17, 1074-1077.
- Ábrahámi, R. A., Kiss, L., Barrio, P., Fülöp, F., Tetrahedron, 2016, 72, 7526-7535.
- Kiss, L., Remete, A. M., Nonn, M., Fustero, S., Sillanpää, R., Fülöp, F., Tetrahedron, 2016, 72, 781-787.
- Kiss, L., Nonn, M., Sillanpää, R., Haukka, M., Fustero, S., Fülöp, F., Chem. Asian J., 2016, 11, 3376-3381.
- Remete, A. M., Nonn, M., Fustero, S., Fülöp, F., Kiss, L., Molecules, 2016, 21, 1493.
- Kiss, L., Petrovszki, Á., Vass, C., Nonn, M., Sillanpää, R., Haukka, M., Fustero, S., Fülöp, F., Chemistry Select, 2017, 2, 3049-3052.
- Remete, A. M., Nonn, M., Fustero, S., Haukka, M., Fülöp, F., Kiss, L., Beilstein J. Org. Chem., 2017, 13, 2364-2371.
- Remete, A. M., Nonn, M., Fustero, S., Haukka, M., Fülöp, F., Kiss, L., Eur. J. Org. Chem., 2018, 27-28, 3735-3742.
- Nonn, M., Fülöp, F., Kiss, L., Fluorine Notes, 2018, 1(116), 1-2, DOI: 10.17677/fn20714807.2018.01.01
- Ouchakour, L., Nonn, M., Kiss, L. Fluorine Notes 2019, 1(122), 1-2, DOI: 10.17677/fn20714807.2019.01.01
- Remete, A. M., Benke, Z., Kiss, L. Fluorine Notes 2019, 6(127), 3-4, DOI: 10.17677/fn20714807.2019.06.02
- Forró, E., Schönstein, L., Kiss, L., Vega-Peñaloza, A., Juaristi, E., Fülöp, F., Molecules, 2010, 15, 3998-4010.
- Musolino, M. G., Cutrupi, C. M. S., Donato, A., Pietropaolo, D., Pietropaolo, R., Appl. Catal. A, 2003, 243, 333-346.
- Musolino, M. G., De Maio, P., Donato, A., Pietropaolo, R., J. Mol. Catal. A: Chem., 2004, 208, 219-224.
- Zsolnai, D., Mayer, P., Szőri, K., London, G., Catal. Sci. Technol., 2016, 6, 3814-3820.
- Dékány, A., Lázár, E., Szabó, B., Havasi, V., Halasi, G.,· Sápi, A., Kukovecz, Á., Kónya, Z.,·Szőri, K., London, G. Catal. Lett., 2017, 147, 1834-1843.
- Sadeghmoghaddam, E., Gu, H., Shon, Y.-S., ACS Catal., 2012, 2, 1838-1845.
- Benedek, G., Palkó, M., Wéber, E., Martinek, T. A., Forró, E., Fülöp, F., Eur. J. Org. Chem., 2008, 21, 3724-3730.
- Wirth, T. (Ed.), Organoselenium Chemistry: Synthesis and Reactions; Wiley-VCH: Weinheim, Germany, 2012.
- Remete, A. M., Novák, T. T., Nonn, M., Haukka, M., Fülöp, F., Kiss, L., Beilstein J. Org. Chem., 2020, 16, 2562-2575.
- Parsons, P. J., Camp, N. P., Edwards, N., Sumoreeah, L. R., Tetrahedron, 2000, 56, 309-315.
- Kiss, L., Forró, E., Sillanpää, R., Fülöp, F., J. Org. Chem., 2007, 72, 8786-8790.
- Coldham, I., Price, K. N., Rathmell, R. E., Org. Biomol. Chem., 2003, 1, 2111-2119.
- Fülöp, F., Palkó, M., Forró, E., Dervarics, M., Martinek, T. A., Sillanpää, R., Eur. J. Org. Chem., 2005, 3214-3220.
- Kanizsai, I., Szakonyi, Z., Sillanpää, R., D'Hooghe, M., De Kimpe, N., Fülöp, F., Tetrahedron: Asymmetry, 2006, 17, 2857.
- Sheldrick, G.M., A short history of SHELX., Acta Cryst., 2008, A64, 112-122.
- Farrugia, L. J. J., Appl. Cryst., 1997, 30, 565.
Статья рекомендована к публикации членом редколлегии д.х.н. С.М. Игумновым
Fluorine Notes, 2021, 134, 5-6


