Поступило в редакцию: Апрель 202
Fluorine Notes, 2020, 129, 5-6
СИНТЕЗ N-МЕТИЛ-N-ПОЛИФТОРАЛКИЛ-D-ГЛЮКАМИНОВ И N-МЕТИЛ-D-ГЛЮКАМИДОВ ФТОРСОДЕРЖАЩИХ КИСЛОТ
Норберт Барис (Norbert Baris), Анико Немес (Anikó Nemes), Денес Сабо (Dénes Szabó), Гитта Шлоссер (Gitta Schlosser), Антал Чампай (Antal Csámpai), Йожеф Рабай (József Rábai )*
*Институт химии, Университет ELTE Eötvös Loránd, Будапешт 1117, Венгрия
E-mail: rabai@elte.hu
Аннотация: Селективное N-алкилирование или N-ацилирование D-глюкамина с использованием (перфторалкил)пропил йодидов/мезилатов или фторированных карбоновых кислот, позволяют синтезировать новые фторированные хиральные расшепляющие агенты.
Ключевые слова: фтор, хиральные третичные амины/амиды, амфифилы на основе углеводов, алкилирование, ацилирование.
Живые организмы обычно синтезируют только один из двух энантиомеров хиральных молекул, однако при их химическом синтезе часто получаются рацемические соединения. Биологическая активность энантиомеров может быть различной или даже противоположной, поэтому энантиомерное разделение необходимо и неизбежно для фармацевтической и тонкой химической промышленности. Строгие правила, введенные в 90-х годах прошлого века в отношении производства и сбыта хиральных лекарств, внесли изменения в стратегию деятельности фармацевтической промышленности [1].
В мире проявляют большой коммерческий интерес к хиральности и стимулированию производства оптически чистых материалов (т. е. отдельных энантиомеров) с помощью методов, применимых для их производства в по крайней мере мультиграммовых количествах , а во многих случаях и в сотнях и тысячах тонн [2].
Существует два основных подхода к получению хиральных соединений: разделение и асимметричный синтез [3]. Несмотря на то, что кристаллизация является ведущим методом разделения диастереомеров [4], некоторые нетрадиционные методы дополняют ее или дают единственные решения для особых случаев [5].
Авторы обнародовали свои результаты по разработке нетрадиционных методов расшепления оптических изомеров, основанных на разделении диастереомерных солей или комплексов с использованием сульфоксидных карбоновых кислот в качестве моделей. Подобные методы включают в себя селективную экстракцию с высаливанием («SOSE») [6, 7] (как один из первых примеров современной энантиоселективной экстракции «жидкость-жидкость» (ELLE) [8]), хироселективный транспорт через жидкие мембраны [7] и стимулируемая нагревом кристаллизация (HFC) [7, 9, 10]. Кроме того, некоторые (CF3)3CO-замещенные фторсодержащие карбоновые кислоты синтезировались и вводились в качестве новых хиральных сольватирующих реагентов для определения энантиомерного соотношения выбранных аминов с помощью ЯМР-спектроскопии 1H и 19F [11].
Разделяющие агенты на основе углеводов играют важную роль в производстве одного определенного энантиомера активных фармацевтических ингредиентов. [3]. Так, (S)-(+)-напроксен непосредственно выделяли из рацемата с высокой энантиомерной чистотой (> 95% е.е.) путем инклюзионной кристаллизации с помощью N-октил-D(-)-глюкамина в качестве хирального «хозяина» [12].
Производство N-полигидроксиалкиламинов, таких как N-метил D-(-)-глюкамин (2), давно известно, такие материалы являются коммерчески доступными. Поверхностно-активные вещества на основе амидов жирных кислот (3) получают реакцией ацилирования незащищенного амина 2O (2) эфиром жирной кислоты (Схема 1) [13 ].
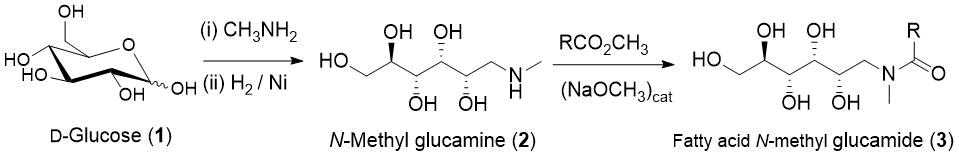
Схема 1. Схема производства глюкамидов жирных кислот.
Эти соединения созданы для использования уникальных воздействий перфторалкильных групп [14] на микроскопические [15] и макроскопические свойства молекул [16]. Здесь мы показываем синтез фторированных хиральных аминов 5a-c и амидов 7a-b, см. Схему 2 и Схему 3 соответственно.
В реакциях алкилирования фторированные алкилиодиды 4a-c [17] реагировали с двукратным молярным избытком N-метил D-глюкамина (2) и избытком K2CO3 в кипящем ацетонитриле в течение 4 ч с целью достижения полной конверсии 4a-c (контроль по ТСХ). После упаривания растворителя избыток (2) удаляли путем растирания сырого продукта в порошок с водой, после чего твердые остатки отфильтровывали, высушивали и перекристаллизовывали из кипящего метанола с получением чистых 3O аминов 5a-c (со средним или высоким выходом в виде белых кристаллов, обладающих узким диапазоном температур плавления). Необходимо отметить, что C8F17(CH2)3OTs был неэффективен для алкилирования (2) в CH3CN с кипячением с обратным холодильником в течение нескольких дней, тогда как был эффективен соответствующий мезилат (см. Схему 2 и раздел «Экспериментальная часть»).
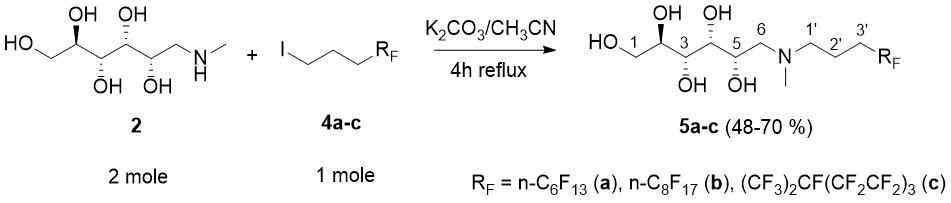
Схема 2 - Получение N-метил-N-[3-(перфторалкил) пропил]-D-глюкаминов 5a-c (Нумерация аминов 5a-c используется для их соотнесения с ЯМР-данными).
Селективное ацилирование N-метил D-глюкамина (2) проводили с использованием протокола для смешанного ангидрида с целью активации 4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-гептадекафторо-ундекановой кислоты 6а, с получением фторсодержащего D-глюкамида 8а с приемлемым выходом после выделения (см. Схему 3). Указанный протокол активации карбоксилом использовался Беном с сотр. (Ben et al.) [18] для синтеза N-метил-N-октаноил-нD-глюкамина 8b.
Однако фторсодержащий амид 8а во время обработки водой создавал плотный гидрогель, который с целью инициирования кристаллизации разрушали добавлением метанола и последующим кипячением (см. раздел «Экспериментальная часть»).
Отметим, что выход D-глюкамида (8b) был увеличен с заявленных в [18] 30% до 79% путем добавления по каплям (2), растворенного в нагретом метаноле, к охлажденному эфирному раствору смешанного ангидрида 7b (см. раздел «Экспериментальная часть»).
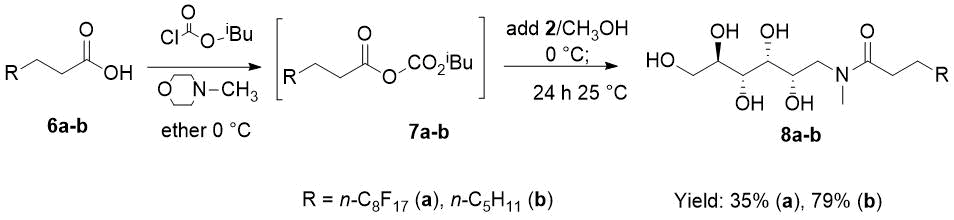
Схема 3. Селективное N-ацилирование N-метил-D-глюкамина (2) в реакционной смеси, образующей смешанные ангидриды 7a-b.
Все новые соединения были охарактеризованы методами 1Н и 13С ЯМР-спектроскопии и масс-спектрометрии с высоким разрешением (HRMS) (см. разделы «Экспериментальная часть», «Supporting Information»). Эти впервые полученные фторированные третичные амины 5a-c и амиды 8a-b будут протестированы в нашей лаборатории для разделения на оптические изомеры выбранных летучих рацемических фторированных соединений.
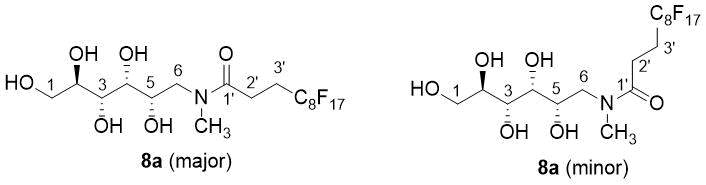
Рисунок 1. Нумерация аминдов 8a(основного и неосновного компонентов) используется для их соотнесения с ЯМР-данными
Следует отметить, что основной фрагмент трет-амина в 5а-с, по-видимому, позволяет ускорять обменные процессы с участием ОН-протонов, поскольку их сигналы объединяются с сигналом от воды в DMSO-d6, используемым в качестве растворителя при ЯМР-измерениях, тогда как в ЯМР 1Н-спектре амида 8а, зарегистрированного с использованием того же растворителя, ОН-сигналы разделяются на дублеты или триплеты (в соответствии с их положением в структуре карбогидрата). С другой стороны, амид 8a был представлен двумя ротамерами или присутствовал в соотношении примерно 2:1 в образце, подвергавшегмся ЯМР-измерениям (см. Рис. 1). Вследствие ярко выраженного эффекта экранирования карбонильного кислорода химический сдвиг 1Н-ЯМР-сигнала от N-СН3 8а (неосновного компонента) заметно уменьшается по сравнению с регистрируемым от амида 8а (основного компонента) (3,02 и 2,83 ppm, соответственно). Несмотря на то, что фторированная цепь не указывалась в качестве объекта в исследуемых продуктах, триплетное расщепление C-3-резонанса явно относилось к прямому присоединению группы CF2.
Экспериментальная часть
Фторированная карбоновая кислота 6а была получена как описано в работах [19] и [20]. CF3CH2OH и фторированные прекурсоры приобретались у компании FC Chemicals, а другие реагенты и органические растворители - у компаний Sigma-Aldrich и Molar Chemicals Kft (Венгрия). ЯМР-спектры 1H- и 13C всех соединений регистрировались в раствора DMSO-d6 в ампулах 5 мм при комнатной температуре на спектрометре Bruker DRX-500 на частотах 500 МГц (1H) и 125 МГц (13C) с использованием сигнала дейтерия в растворителе в качестве дейтериевого стабилизатора, и тетраметилсилана (TMS) – в качестве внутреннего стандарта. Спектры HSQC, HMBC и COSY, которые обеспечивают точную идентификацию ЯМР-сигналов 1H и 13C, были получены с использованием стандартных импульсных программ компании Bruker.
Определение молекулярной массы и получение тандемного масс-спектра соединений проводилось методом масс-спектрометрии с ионизацией электрораспылением (ESI-MS) на масс-спектрометре с ионной ловушкой модели Bruker Daltonics Esquire 3000 plus (Германия). Точки плавления этих соединений определялись на аппаратуре для микроизмерений температуры плавления производства компании Boetius и не корректировались. Контроль реакций производился методом тонкослойной хроматографии (TLC) (с использованием силикагеля 60 F254, Merck Darmstadt). Оптическое вращение измерялось с помощью поляриметра Polamat A, Zeiss, Jena (концентрация указывалась в г/100 мл).
(2R, 3R, 4R, 5S) -6 - [(метил (4,4,5,5,6,6,7,7,8,8,9,9,9-тридекафторнонил)амино]гексан-1,2,3,4,5-пентаол (5а)
Смесь N-метил-глюкамина (2, 4,29 г, 22 ммоль), 3-(перфторгексил)пропилиодида (4а, 5,37 г, 11 ммоль) и K2CO3 (3,04 г, 22 ммоль) кипятили в ацетонитриле (180 мл) в течение 4 ч при интенсивном перемешивании. Растворитель удаляли в вакууме, затем полученный остаток перемешивали с водой (40 мл) в течение 1 ч, фильтровали, промывали водой и высушивали. Неочищенный продукт кристаллизовали из MeOH (16 мл) для получения чистого амина 5а (3,75 г, 61,5%) в виде белых кристаллов с температурой плавления 91‑93 °С.
[α]578 = – 4.03; [α]546 = – 4.36; [α]436 = – 6.38; [α]406 = –6.71 (c = 3.0 DMF)
1H NMR (DMSO-d6): 3.67 (m, 1H, H-5); 3.58 (m, 1H, H-4); 3.56 (dd, J=10.8 Hz and 3.8 Hz, 1H, H-1A); 3.48 (m, 1H, H-2); 3.43 (dd, J=7.6 Hz and 2.0 Hz, 1H, H-3); 3.38 (dd, J=10.8 Hz and 5.8 Hz, 1H, H-1B); 2.44-2.39 (перекрывающиеся m, 3H, H-1’ and H-6A); 2.33 (dd, J=13.2 Hz and 6.8 Hz, 1H, H-6B); 2.20 (m, 2H, H-3’); 2.16 (s, 3H, N-CH3); 1.63 (qui, J=7.9 Hz, 2H, H-2’).
13C NMR (DMSO-d6) (нефторированные атомы углерода: 72.9 (C-3); 72.2 (C-2); 70.9 (C-4); 70.8 (C-5); 64.0 (C-1); 60.7 (C-6); 56.9 (C-1’); 42.6 (N-CH3);28.4 (t, J=21.2 Hz, C-3’); 18.2 (C-2’).
HRMS (ESI) m/z: calcd. for C16H22F13NO5555.12904; found 555.12721. Mass error: –3.3 ppm.
(2R, 3R, 4R, 5S) -6 - [(4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-гептадекафтороундецил)(метил амино гексан-1,2,3,4,5-пентаол (5b)
Метод А:
Смесь N-метилглюкамина (2, 5,85 г, 30 ммоль), 3-(перфтороктил)пропилиодида (4b, 8,85 г, 15 ммоль) и K2CO3 (4,14 г, 30 ммоль) кипятили в ацетонитриле (300 мл) в течение 4 ч при интенсивном перемешивании. Растворитель удаляли в вакууме, затем остаток перемешивали с водой (50 мл) в течение 1 ч, фильтровали, промывали водой и высушивали. Неочищенный продукт кристаллизовали из МеОН (35 мл) для получения чистого амина 5b в виде белых кристаллов (Fw = 655, 6,33 г, 64,5%) с температурой плавления 103‑105 °С. При использовании 30 ммоль 4b получали 13,74 г (70%) амина 5b в виде белых кристаллов с температурой плавления 104-107 °С.
[α]578 = − 3.07; [α]546 = − 3.41; [α]436 = − 6.48; [α]406 = − 6.83 (c = 3, DMF).
1H NMR (DMSO-d6): 3.63 (m, 1H, H-5); 3.54 (m, 1H, H-4); 3.52 (dd, J=10.8 Hz and 3.8 Hz, 1H, H-1A); 3.43 (m, 1H, H-2); 3.36 (dd, J=7.6 Hz and 2.0 Hz, 1H, H-3); 3.32 (dd, J=10.8 Hz and 5.8 Hz, 1H, H-1B); 2.42-2.33 (перекрывающиеся m, 3H, H-1’ and H-6A); 2.27 (dd, J=13.2 Hz and 6.8 Hz, 1H, H-6B); 2.16 (m, 2H, H-3’); 2.12 (s, 3H, N-CH3); 1.58 (qui, J=7.9 Hz, 2H, H-2’).
13C NMR (DMSO-d6) (нефторированные атомы углерода: 72.6 (C-3); 71.9 (C-2); 70.7 (C-5); 70.6 (C-4); 63.9 (C-1); 60.6 (C-6); 56.8 (C-1’); 42.6 (N-CH3); 28.1 (t, J=21.5 Hz, C-3’); 18.0 (C-2’).
HRMS (ESI) m/z: calcd. for C18H22F17NO5 655.12265; found 655.12024. Mass error: -3.7 ppm.
Метод Б:
Смесь 3-(перфтороктил)пропилмезилата (0,56 г, 1,0 ммоль), N-метилглюкамина (2, 0,39 г, 2 ммоль) и K2CO3 (0,28 г, 2 ммоль) в ацетонитриле (10 мл) в течение 1 ч перемешивали и в течение 24 ч нагревали при температуре 70 °С в запаянном сосуде, затем растворитель удаляли упариванием в вакууме с использованием роторного испарителя. Твердый остаток обрабатывали водой (15 мл) и фильтровали. Неочищенный продукт отфильтровывали под вакуумом и высушивали, получая при этом 0,40 г (62%) белых кристаллов с температурой плавления 102-104 °C.
HRMS (ESI) m/z: calcd. for C18H22F17NO5 655.12265; found 655.12028. Mass error: -3.6 ppm.
(2R,3R,4R,5S)-6-{[4,4,5,5,6,6,7,7,8,8,9,9,10,11,11,11-hexadecafluoro-10-(трифторметил) ундецил](метил)амино} гексан-1,2,3,4,5-пентаол (5с);
Смесь N-метил-глюкамина (2, 7,0 г, 36 ммоль), 3-(изоперфторононил)пропилиодида (4с, 11,6 г, 18 ммоль) и K2CO3 (4,98 г, 36 ммоль) кипятили в ацетонитриле (350 мл) в течение 4 ч при интенсивном перемешивании. Растворитель удаляли в вакууме, затем остаток перемешивали с водой (60 мл) в течение 1 ч. К полученной водной суспензии добавляли МеОН (130 мл), смесь нагревали до получения прозрачного раствора. После охлаждения осадок отфильтровывали, сушили и кристаллизовали из МеОН (45 мл), получая чистый амин 5с (6,1 г, 48%) с температурой плавления 86-91 °С.
[α]578 = −3.07; [α]546 = −3.41; [α]436 = −6.48; [α]406 = −6.83. (c = 1, CF3CH2OH).
1H NMR (DMSO-d6): 3.67 (m, 1H, H-5); 3.58 (m, 1H, H-4); 3.56 (dd, J=10.8 Hz and 3.8 Hz, 1H, H-1A); 3.47 (m, 1H, H-2); 3.40 (dd, J=7.6 Hz and 2.0 Hz, 1H, H-3); 3.35 (dd, J=10.8 Hz and 5.8 Hz, 1H, H-1B); 2.46-2.37 (перекрывающиеся m’s, 3H, H-1’ and H-6A); 2.31 (dd, J=13.2 Hz and 6.8 Hz, 1H, H-6B); 2.17 (m, 2H, H-3’); 2.15 (s, 3H, N-CH3); 1.62 (qui, J=7.9 Hz, 2H, H-2’).
13C NMR (DMSO-d6) (нефторированные атомы углерода: 72.7 (C-3); 71.9 (C-2); 70.7 (C-4); 70.6 (C-5); 63.9 (C-1); 60.6 (C-6); 56.8 (C-1’); 42.6 (N-CH3); 28.1 (t, J=21.5 Hz, C-3’); 18.0 (C-2’).
HRMS (ESI) m/z: calcd. for C19H22F19NO5 705.11946; found 705.11733. Mass error: -3.0 ppm.
4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,1-гептадекафтор-N-метил-N -[(2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил]ундеканамид (8a)
Раствор 3-перфтороктилпропановой кислоты 6а (4,92 г, 10 ммоль) в диэтиловом эфире (20 мл) охлаждали до 0 °С и прибавляли в него изобутилхлорформиат (1,37 г, 10 ммоль). После перемешивания в течение 10 мин в смесь добавляли N-метилморфолин (1,01 г, 10 ммоль) и перемешивали еще в течение 10 мин. Осажденную хлористоводородную соль удаляли методом фильтрования, затем в фильтрат по каплям в течение 10 мин добавляли нагретый раствор (50°С) N-метилглюкамина (0,88 г, 4,5 ммоль) в МеОН (30 мл). Смесь перемешивали в течение ночи при комнатной температуре, затем растворитель удаляли в вакууме. Остаток растирали с диэтиловым эфиром, а кристаллы фильтровали под вакуумом и сушили, получая (2,6 г) неочищенного продукта, который перекристаллизовывали из МеОН (10 мл), получая при этом чистый амид 8a (1,04 г, 35%) с температурой плавления 95-210 °С. Это уникальный диапазон температур плавления может быть обусловлено изменениями при нагревании структуры H-связанной сети в твердом состоянии. Необходимо отметить, что в растворе (DMSO-d6) наличие двух разных ротамеров идентифицировалось ЯМР-методом (см. "Supporting Information").
1H NMR (DMSO-d6): Наиболее диагностически важные и однозначные определения без указания интенсивности для амида (major): 4.94 (d, J=5.0 Hz, 5-OH); 4.34 (t, J=5.5 Hz, 1-OH); 3.77 (m, H-5); 3.56 (m, H-1A);3.37 (m, H-1B); 3.42-3.27 (overlapping m’s, H-6A and H-6B in the major and minor components); 2.83 (s, N-CH3); 2.62 (m, H-3’).
13C NMR (DMSO-d6) Наиболее диагностически важные и однозначные определения без указания интенсивности для амида 8a (major): 170.16 (C-1’); 70.5 (C-5); 63.78 (C-1); 52.3 (C-6);34.1 (N-CH3); 26.5 (t, J=21.3 Hz, C-3’); 23.8 (C-2’).
1H-ЯМР (DMSO-d6): Наиболее диагностически важные и однозначные определения без указания интенсивности для амида 8a (minor): 4,69 (d, J = 5,0 Гц, 5-OH); 4,30 (t, J = 5,5 Гц, 1-OH); 3.42-3.27 (перекрывающиеся m, H-6A и H-6B major и minor); 3,02 (s, N-CH3); 2,80 (m, Н-2’).
13C-ЯМР (DMSO-d6): Наиболее диагностически важные и однозначные определения без указания интенсивности для амида 8a (minor): 170,06 (C-1’); 71,5 (С-5); 63,75 (С-1); 51,6 (С-6); 36,9 (N-CH3); 26,3 (t, J = 21,9 Гц, C-3’); 24,2 (С-2’).
HRMS (ESI) m/z: calcd. for C18H20F17NO6 669.10192; found 669.10150. Mass error: -6.0 ppm.
N-метил-N-((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил) октанамид (8b)
К охлажденному на льду раствору октановой кислоты (2,88 г, 20 ммоль) в эфире (30 мл) добавляли ClCOOiBu (2,73 г, 20 ммоль), смесь перемешивали в течение 10 мин. После добавления N-метилморфолина (2,02 г, 20 ммоль) осажденный гидрохлорид удаляли методом фильтрования. Фильтрат охлаждали льдом; при этом по каплям добавляли нагретый до 40-50°C раствор N-метилглюкамина (1,75 г, 9 ммоль) в 60 мл эфира. После этого смеси давали возможность нагреться до комнатной температуры и оставляли на ночь. Растворитель упаривали, а оставшееся масло обрабатывали небольшим объемом эфира и растирали для инициирования кристаллизации. Отфильтровывали 2,60 г (81%) твердого вещества, что при перекристаллизации из ацетонитрила (10 мл) дает 2,28 г (79%) белых кристаллов с температурой плавления 84-85 °С.
MS (EI) m/z: calcd for C15H31NO6 321.2; observed 322.4 (M+H)+.
Благодарности
Авторы выражают благодарность Национальному бюро исследований, разработок и инноваций (K115764) за поддержку данной работы. Гитта Шлоссер выражает признательность за свою поддержку грантом на исследования от МТА Яноша Боляй (János Bolyai) и в рамках расширенной исследовательской МТА-программы для докторантуры Венгерской академии наук (HAS, MTA).
Информация по ЯМР спектрам соединений представлена в PDF версии статьи.
Список литературы
- E. Pálovics, Zs. Szeleczky, B. Fődi, F. Faigl, E. Fogassy, Prediction of the efficiency of diastereoisomer separation on the basis of the behavior of enantiomeric mixtures, RSC Adv., 2014, 4, 21254-21261. DOI: 10.1039/C4RA00526K
- N. A. Vaidya, Diastereomeric crystallisation – the ‘classical’ chiral technology; Innovations in Pharmaceutical Technology (2000) 82-85; accessed at 01 December, 2019. http://iptonline.com/articles/public/IPTNINE82NoPrint.pdf
- D. J. Ager (Ed.), Handbook of chiral chemicals. 2nd ed. ©, 2006, Taylor & Francis Group, https://doi.org/10.1201/9781420027303
- Pálovics, E.; Faigl, F., Fogassy, E., Chap.1. Separation of the Mixtures of Chiral Compounds by Crystallization. Advances in Crystallization Processes, Yitzhak Mastai (Ed.), InTech, 2012. DOI: 10.5772/33592
- E. Fogassy, M. Nógrádi, E. Pálovics, J. Schindler, Resolution of Enantiomers by Non-Conventional Methods. Synthesis, 2005, 1555-1568. DOI: 10.1055/s-2005-869903
- J. Rábai, Salting Out Selective Extraction – A Novel Method for the Optical Resolution of Chiral Sulfinylcarboxylic Acids and Its Application for the Convenient Determination of Optical Purity, Angew. Chem., Int. Ed., 1992, 31, 1631-1633. https://doi.org/10.1002/anie.199216311
- A. Nemes, D. Szabó, J. Rábai, Comparison of resolution methods for racemic 8‑(phenylsulfinyl)-1-naphthoic acid, Tetrahedron: Asymmetry, 2017, 1078-1082. DOI: 10.1016/j.tetasy.2017.07.001
- B. Schuur, B. J. V. Verkuijl, A. J. Minnaard, J. G. de Vries, H. J. Heeres and B. L. Feringa, Chiral separation by enantioselective liquid–liquid extraction, Org. Biomol. Chem., 2011, 9, 36-51. DOI: 10.1039/C0OB00610F
- D. Szabó, A. Nemes, I. Kövesdi, V. Farkas, M. Hollósi, J. Rábai, Synthesis and characterization of fluorous (S)- and (R)-1-phenylethylamines that effect heat facilitated resolution of (±)-2-(8-carboxy-1-naphthylsulfinyl)benzoic acid via diastereomeric salt formation and study of their circular dichroism, J. Fluorine Chem., 2006, 127, 1405-1414. https://doi.org/10.1016/j.jfluchem.2006.05.011
- A. Nemes, E. Vass, I. Jalsovszky, D. Szabó, Synthesis of enantiopure 2‑iodomandelic acid and determination of its absolute configuration by VCD spectroscopy, Chem. Pap., 2019, 73, 47-54. https://doi.org/10.1007/s11696- 018-0568-6
- A. Nemes, T. Csóka, Sz. Béni, V. Farkas, J. Rábai, D. Szabó, Chiral recognition studies of α-(nonafluoro-tert- butoxy)carboxylic acids by NMR spectroscopy, J. Org. Chem., 2015, 80(12), 6267-6274. https://doi.org/10.1021/acs.joc.5b00706
- Xuejun Yuan, Jiguo Li, Yunqi Tian, Gene-Hsiang Lee, Xie-Ming Peng, Rongguang Zhu and Xiaozeng Youa, Efficient resolution of naproxen by inclusion crystallization with N-octyl-glucamine and structure characterization of the inclusion complex, Tetrahedron: Asymmetry, 2001, 12, 3015–3018. https://doi.org/10.1016/S0957- 4166(01)00536-5
- (a) J. J. Scheibel, D. S. Connor, R. E. Shumate, and J. C. T. R. B. St. Laurent, Process for Preparing N-Alkyl Polyhydroxy Amines and Fatty Acid Amides Therefrom in Hydroxy Solvents, EP 0558515B, 1990, Procter & Gamble, Chem. Abstr. 1992, 117, 114045; (b) K. Hill and C. LeHen-Ferrenbach, Sugar-Based Surfactants for Consumer Products and Technical Applications, Chapter 1, In: Cristóbal Carnero Ruiz (Editor), Sugar-Based Surfactants: Fundamentals and Applications, CRC Press Tayrol & Francis Group, Boca Raton, FL, 2009.
- (a) I. T. Horváth, J. Rábai, Facile Catalyst Separation without Water: Fluorous Biphase Hydroformylation of Olefins, Science, 1994, 266, 72-75, DOI:10.1126/science.266.5182.72; (b) Handbook of Fluorous Chemistry, J. A. Gladysz, D. P. Curran, I. T. Horváth (Editors), Wiley/VCH: Weinheim, 2004, DOI: 10.1002/3527603905; (c) Fluorous Chemistry, Volume Editor: I. T. Horváth, Topics in Currant Chemistry, Springer, 2012, 308, Heidelberg. DOI 10.1007/978-3-642-25234-1; (d) J.-M. Vincent, M. Contel, G. Pozzi, R. H. Fish, How the Horváth paradigm, Fluorous Biphasic Catalysis, affected oxidation chemistry: Successes, challenges, and a sustainable future, Coord. Chem. Rev., 2019, 380, 584-599. https://doi.org/10.1016/j.ccr.2018.11.004
- M. Cametti, B. Crousse, P. Metrangolo, R. Milani, G. Resnati, The fluorous effect in biomolecular applications, Chem. Soc. Rev., 2012, 41, 31–42. DOI: 10.1039/C1CS15084G
- M. P. Kraft, Fluorocarbons and fluorinated amphiphiles in drug delivery and biomedical research, Adv. Drug Delivery Rev., 2001, 47, 209-228. DOI: 10.1016/s0169-409x(01)00107-7
- B. Menczinger, G. Jakab, D. Szabó and J. Rábai, Synthesis of 1-iodo-3-perfluoroalkylpropanes and 1-iodo-4- perfluoroalkylbutanes, Fluorine notes, 2014, 94, 7-8. http://notes.fluorine1.ru/public/pdfs/94_4.pdf
- C. J. Capicciotti, M. Leclère, F. A. Perras, D. L. Bryce, H. Paulin, J. Harden, Y. Liu, R. N. Ben, Potent Inhibition of Ice Recrystallization by Low Molecular Weight Carbohydrate-Based Surfactants and Hydrogelators, Chem. Sci., 2012, 3, 1408-1416. DOI: 10.1039/c2sc00885h.
- T. Miura, Y. Hirose, M. Ohmae, and T. Inazu, Fluorous Oligosaccharide Synthesis Using a Novel Fluorous Protective Group, Org. Lett., 2001, 3, 3947-3950. DOI: 10.1021/ol016838o.
- B. Hungerhoff, H. Sonnenschein, F. Theil, Combining Lipase-Catalyzed Enantiomer-Selective Acylation with Fluorous Phase Labeling: A New Method for the Resolution of Racemic Alcohols, J. Org. Chem., 2002, 67, 1781-1785. https://doi.org/10.1021/jo010767p
Статья рекомендована к публикации членом редколлегии д.х.н. С.М. Игумновым
Fluorine Notes, 2020, 129, 5-6


