Поступило в редакцию: Декабрь 2019
Fluorine Notes, 2019, 127, 3-4
СТЕРЕОКОНТРОЛИРУЕМЫЙ СИНТЕЗ ФТОРСОДЕРЖАЩИХ ФУНКЦИОНАЛИЗИРОВАННЫХ ПРОИЗВОДНЫХ β-ЛАКТАМА С ПОМОЩЬЮ РЕАКЦИЙ КРОСС-МЕТАТЕЗИСА
Аттила М. Ремете (Attila M. Remete)1,2, Жанетт Бенке (Zsanett Benke)1,2, Лоранд Кисс (Loránd Kiss)1,2*
1Институт фармацевтической химии, Сегедский университет, H-6720 Szeged, Eötvös u. 6, Венгрия
2 Университет Сегеда, Межотраслевой Центр передового опыта, Институт фармацевтической химии, H-6720 Szeged, Eötvös u. 6, Венгрия
Е-mail: kiss.lorand@pharm.u-szeged.hu
Аннотация: К функционализированным β-лактамам и их производным в последние десятилетия в медицинской химии проявляется все больший интерес. В связи с их высоким фармакологическим потенциалом химия фторированных β-лактамов и β-аминокислот считается бурно расширяющейся областью исследований. Стереоконтролируемый синтез различных фторсодержащих ацильных β-лактамов производился из некоторых легкодоступных ненасыщенных бициклических β-лактамных изомеров. Стратегия синтеза при этом состояла в метатезисе с раскрытием кольца ненасыщенных бициклических азетидинонов с последующим кросс-метатезисом со фторсодержащими олефинами. Кросс-метатезисные преобразования, выполненные при различных условиях с целью изучения хемодифференциации олефиновых связей, привели к получению соответствующих функционализированных производных β-лактама.
Ключевые слова: β-лактам, раскрытие кольца, функционализация, метатезис, селективность, фтор.
Теоретическое введение
Изучению азетидин-2-онов или β-лактамов в органической и медицинской химии уделяется большое внимание. Эти соединения проявляют широкий спектр биологической активности. Так, β-лактамные антибиотики относят к числу наиболее важных антибактериальных средств [1, 2]. В других работах сообщалось об ингибировании протеазы, абсорбции холестерина, синтезы жирных кислот человека, а также о противоопухолевых свойствах этих антибиотиков [3, 4]. β-Лактамы также являются ценными полупродуктами для синтеза гетероциклов, β-аминокислот, пептидов, таксоидов и других соединений [5-8].
В связи с большим значением фторированных биомолекул для разработки лекарственных средств, введение одного или нескольких атомов фтора в органическую молекулу вызывало большой интерес в течение последних десяти лет [9 - 13]. Благодаря ценности β-лактамов этот интерес также распространялся и на фторсодержащие азетидин-2-оны, которые часто синтезируются с использованием фторированного реагента во время гетероциклизации [14 - 19], однако также может применяться и фторирование на поздней стадии (т. е. введение фтора после синтеза азетидинонового скелета) [20]. β-Лактамы имеют большое значение при синтезе различных фторированных молекул, например, конъюгатов сахаров и β-аминокислот [15], амидоэфиров [18], аминопропанов и гетероциклов [17], а также таксоидов и β-аминокислот [20].
Соответствующая биологическая активность была зарегистрирована для фтор-таксоидов [20] и некоторых N-арил-3,3-дифторазетидин-2-онов [21].
Одним из важнейших достижений в области синтеза за последние два десятилетия стало появление реакций метатезиса олефинов. Этот прорыв стал возможен благодаря коммерчески доступным высокоактивным Ru-катализаторам, не затрагивающим различные функциональные группы (общепринятые обозначения: G1 - для 1-го поколения катализаторов Граббса, G2 - для 2-го поколения катализаторов Граббса, HG1 - для 1-го поколения катализаторов Ховейды–Граббса, и HG2 - для 2-го поколения катализаторов Ховейды–Граббса) [22, 23]. Этот перспективный новый способ позволил синтезировать соединения, которые ранее было трудно или невозможно получить [24, 25].
Обсуждение результатов
В наших недавних исследованиях нами с помощью метода метатезисного раскрытия кольца /кросс-метатезиса (ROM/CM)) функционализированные моноциклические азетидиноны были успешно преобразованы в ненасыщенные бициклические β-лактамы [26, 27]. В настоящее время целью наших исследований является расширение этого стереоконтролируемого направления до синтеза фторсодержащих β-лактамов с использованием фторированных олефинов на стадии СМ-реакции.
Первым из выбранных нами субстратов был бициклический лактам (±)-1, полученный из 1,5-циклооктадиена. Дециклизация этиленом осуществлялась в соответствии с работой [26]. Функционализация полученного таким методом алкенилзамещенного лактама (±)-2 предпринималась с использованием трех различных коммерчески доступных высокофторированных алкенов. Реакции кросс-метатезиса в присутствии четырех коммерчески доступных катализаторов (G1, G2, HG1, HG2) с 2-бром-3,3,3-трифтор-1-пропеном и 4-бром-3,3,4,4-тетрафтор-1-бутеном не удались, но реакция с аллил 1,1,2,3,3,3-гексафторпропиловым эфиром оказалась эффективной. Максимальный выход продукта двойного присоединения (±)-3 был получен с катализатором типа HG2 (см. схему 1).
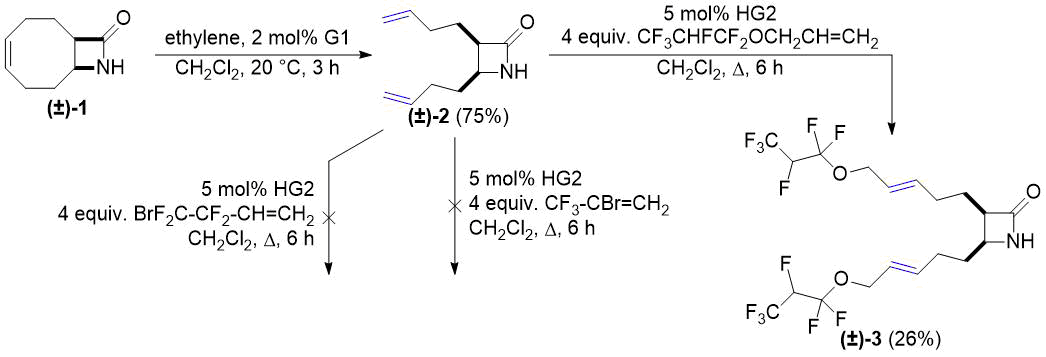
Схема 1. Кросс-метатезис лактама (±)-2 с фторированными алкенами.
Соединение (±)-4, следующий выбранный нами субстрат, было получено путем N-Boc-защиты лактама (±)-1. Модификация описанной в работе [28] методики привела к увеличению выхода продукта на этой стадии. ROM соединения (±) -4 выполнялось аналогично дециклизации (±)-1. Продукт (±)-5, полученный с выходом 77%, затем подвергали СМ-реакции в присутствии катализатора HG2. Аналогично кросс-метатезису (±)-2, реакция с аллил 1,1,2,3,3,3-гексафторпропиловым эфиром прошла успешно и привела к получению соединения (±)-6, однако оказалась неудачной с 2-бром-3,3,3-трифтор-1-пропеном и 4-бром-3,3,4,4-тетрафтор-1-бутеном (см. схему 2).
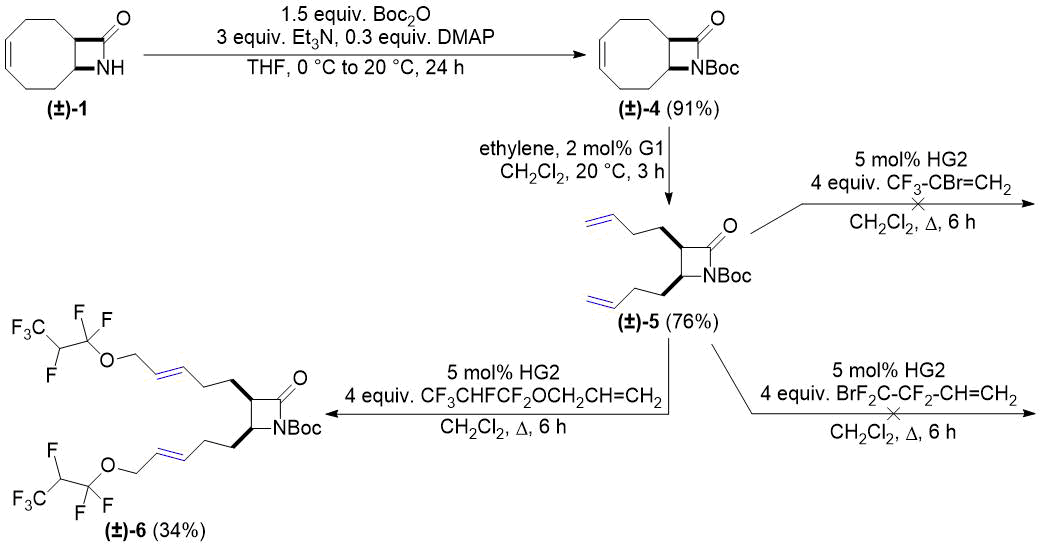
Схема 2. Кросс-метатезис Boc-защищенного лактама (±)-5 с фторированными алкенами.
Нашу работу мы продолжили с бициклическим лактамом (±)-7 (региоизомер лактама (±)-1), полученным из 1,3-циклооктадиена. Раскрытие этого цикла этиленом осуществлялось методом, ранее описанным в работе [26]. С учетом СМ-реакций в схемах 1 и 2, функционализация полученного продукта (±)-8 предпринималась только с аллил-1,1,2,3,3,3-гексафторпропиловым эфиром. Преобразование, выполненное в присутствии катализатора HG2, привело к образованию двусвязного продукта (±)-9 с выходом 35% (см. схему 3).
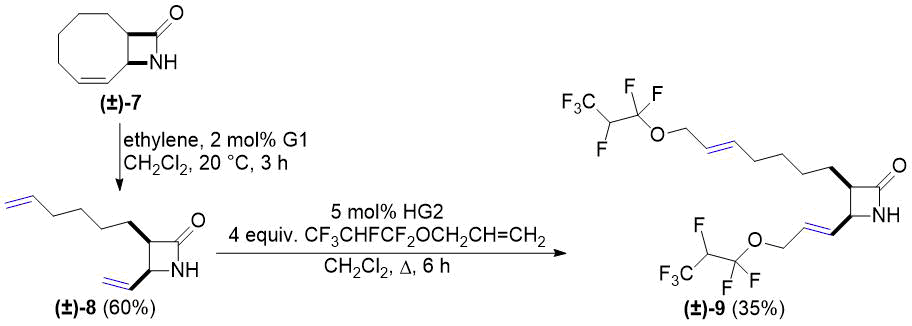
Схема 3. Кросс-метатезис лактама (±) -8 с фторированным алкеном.
После этого мы синтезировали бициклический лактам (±)-10 (N-Boc-защищенное производное (±)-7). Его реакция с этиленом протекала аналогично реакции для соединений (±)-1 и (±)-4. Кросс-метатезис полученного таким образом (±)-11 с аллилом 1,1,2,3,3,3-гексафторпропилового эфира приводил не только к образованию продукта двойного метатезиса (±)-12, но и к образованию моно-продукта (±)-13 (см. схему 4).
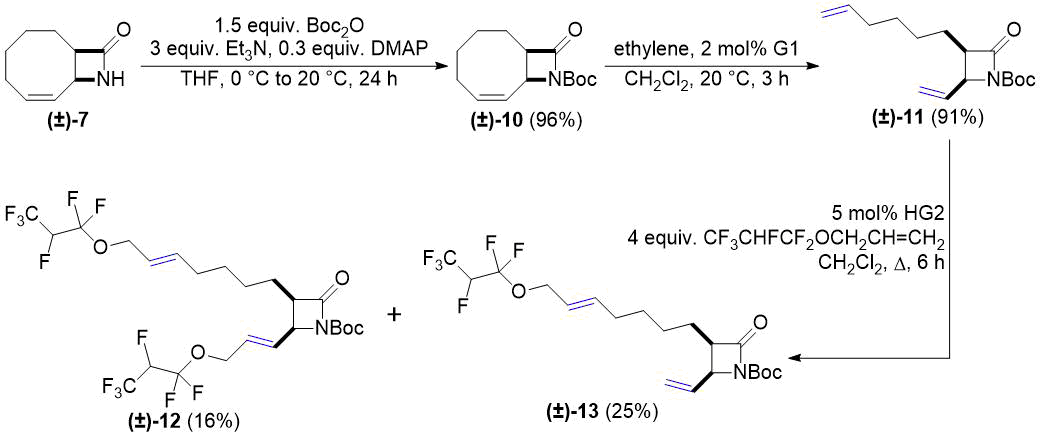
Схема 4. Кросс-метатезис лактама (±)-11 с фторированным алкеном.
Получению такого конечного результата этой реакции могут способствовать два фактора. Прежде всего, стало известно, что хелатирование промежуточного соединения металлоцикла с карбонильными кислородами препятствует дальнейшим реакциям из-за стабилизации металлоцикла [29-31]. При метатезисе винильной группы, присоединенной непосредственно к лактамному кольцу (±)-11, возможно образование шестичленного хелатного кольца с Boc-оксогруппой (см. схему 5, (±)- T2 (±) - T3). В результате реакционная способность связи C=C уменьшается. Другим вероятным фактором является пространственное затруднение этой винильной группы, присоединенной к сильно замещенному азетидиноновому кольцу, что замедляет ее реакцию с Ru-алкилиденовым катализатором (см. схему 5).
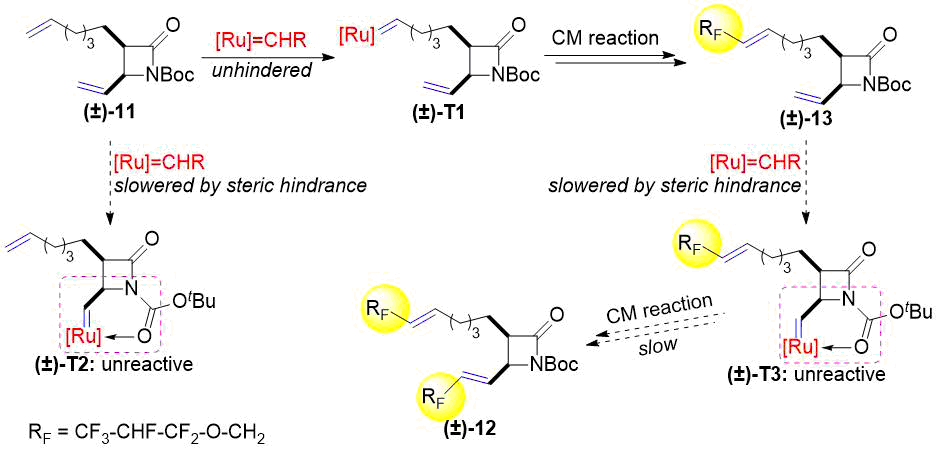
Схема 5. Образование продукта монометатезиса (±)-13 и продукта двойного метатезиса(±)-12.
В дальнейшем лактам (±)-14 и его N-Boc-защищенное производное (±)-19 подвергались ROM в соответствии с нашими предыдущими работами [26, 27]. Кросс-метатезис аллил 1,1,2,3,3,3-гексафторпропилового эфира с соединениями (±)-15 и (±)-20 приводит к температурно-зависимым результатам (см. схемы 6 и 7). При кипячении с обратным холодильником в CH2Cl2 соединений (±)-15 и (±)-20 получают соответствующие продукты двойного метатезиса (±)-16 и (±)-21. При комнатной температуре, в свою очередь, оба соединения (±)-15 и (±)-20 превращались в смеси с соотношением 1,5:1 двух моно-продуктов. Этот результат стал аналогичным результатам наших предыдущих исследований хемоселективности реакций кросс-метатезиса (см. работы [26, 27]). К сожалению, попытки разделения смесей продуктов в обоих случаях оказались неудачными.
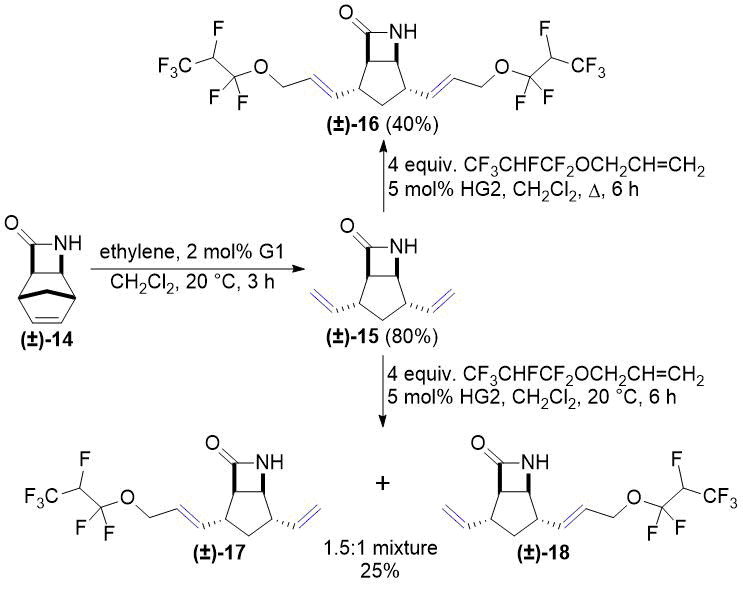
Схема 6. Кросс-метатезис лактама (±)-15 с фторированным алкеном.
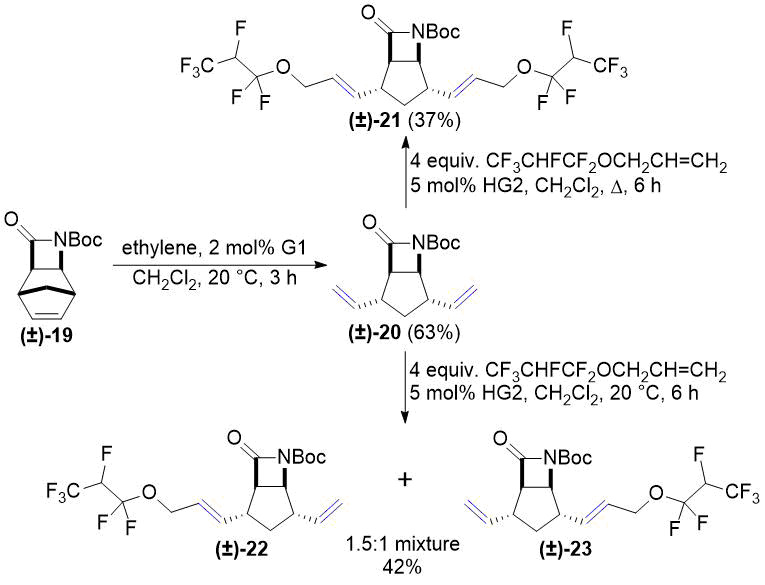
Схема 7. Кросс-метатезис лактама (±)-20 с фторированным алкеном.
Выводы
В настоящей работе описан подход к синтезу новых β-лактамов, содержащих атомы фтора в своих заместителях, и образующихся в реакциях кросс-метатезиса (СМ). Концепция синтеза включала в себя преобразование различных β-лактамов с двумя олефиновыми заместителями с помощью СМ-реакции со фторсодержащими алкеновыми реагентами. Реакция кросс-метатезиса β-лактамов с двумя различными олефиновыми заместителями, в зависимости от их структуры, дает интересные моно- или ди- производные. Стереохимические факторы, обусловленные образованием хелатного кольца дают правдоподобное объяснение конечному результату СМ-реакций. .В настоящее время в наших лабораториях проводят дальнейшие исследования по синтезу новых фторзамещенных β-лактамов с помощью СМ-реакций.
Экспериментальная часть
Общая информация
Химические вещества приобретались у компании Sigma-Aldrich. Растворители использовались в том виде, в каком они были получены от поставщиков. Температура плавления определялась с помощью измерителя Kofler. Элементный анализ проводился с помощью анализатора элементов типа Perkin–Elmer CHNS-2400 Ser II. Силикагель 60 F254 приобретался у компании Merck. ЯМР-спектры регистрировались при комнатной температуре на спектрометре Bruker Avance 400 (1H-частота - 400.13 МГц; 19F-частота - 376.50 МГц, 13C-частота - 100.76 МГц) или на спектрометре типа Bruker Avance Neo (1H-частота - 500.20 МГц; 19F-частота - 470,66 МГц, 13C-частота - 125,78 МГц) с CDCl3 или d6-DMSO в качестве растворителя, используя сигнал от дейтерия в растворителе для стабилизации поля. Химические сдвиги для 1Н и 13С приведены относительно ТМС, а для 19F - относительно CFCl3 (0.00 м.д.).
Общая методика метатезисного раскрытия кольца
500 мг β-Лактама растворяли в 100 мл безводного CH2Cl2, а затем в эту смесь вводили катализатор Граббса 1-го поколения (2 мол. %) и перемешивали при температуре 20 °C в атмосфере этилена в течение трех часов (контроль по ТСХ). После завершения реакции добавляли 0.3 г раствора NaHCO3, 25 мл H2O и 5 мл EtOH, полученную смесь перемешивали при температуре 20 °C еще в течение 2 ч для деактивации катализатора. Затем добавляли 30 мл H2O, и экстрагировали CH2Cl2 (3×40 мл). Органический слой высушивали над Na2SO4 и концентрировали при пониженном давлении. Сырое вещество очищалось с помощью колоночной хроматографии на силикагеле (н-гексан/EtOAc или н-гексан/ацетон).
Общая методика кросс-метатезиса
150 мг β-Лактама растворяли в 30 мл безводного CH2Cl2, а затем вводили в этот раствор катализатор Ховейды-Граббса 2-го поколения (5 мол. %) и 4 эквив. 3-(1,1,2,3,3,3-гексафторпропоксил)проп-1-ена. Реакционную смесь перемешивали при температуре 40 °C в атмосфере аргона в течение 6 ч и контролировали методом тонкослойной хроматографии. После завершения реакции добавляли 0,3 г раствора NaHCO3, 25 мл H2O и 5 мл EtOH, полученную смесь перемешивали при температуре 20 °C еще в течение 2 ч для деактивации катализатора. Затем добавляли 20 мл H2O, и экстрагировали CH2Cl2 (3×40 мл). Органический слой высушивали над Na2SO4 и концентрировали при пониженном давлении. Сырое вещество очищалось с помощью колоночной хроматографии на силикагеле (н-гексан/EtOAc или н-гексан/ацетон).
Общая методика синтеза N-Boc-защищенных лактамов
200 мг β-Лактама растворяли в 40 мл ТГФ, а затем при температуре 0 °C добавляли 3 экв. триэтиламина, 1.5 экв. ди-трет-бутилдикарбоната и 0.3 экв. 4-диметиламинопиримидина. Реакционную массу перемешивали в течение 1 ч, затем нагревали до комнатной температуры и перемешивали еще в течение 24 ч. После того, как тонкослойная хроматография показала завершение реакции, добавляли 30 мл H2O, затем экстрагировали с помощью EtOAc (3×20 мл). Органический слой высушивали над Na2SO4 и концентрировали при пониженном давлении. Сырое вещество очищалось с помощью колоночной хроматографии на силикагеле (н-гексан/EtOAc 10:1).
Для определения характеристик новых соединений и оценки ЯМР-данных см. дополнительную информацию.
Благодарности
Авторы выражают благодарность Венгерскому исследовательскому фонду за финансовую поддержку проекта NKFIH № K 119282, а также за финансовую поддержку проекта GINOP-2.3.2-15-2016-00038. Эти исследования поддерживались также субсидированным ЕС грантом EFOP-3.6.1-16-2016-00008 Министерства людских ресурсов Венгрии.
Список литературы
- Kong, K.-F.; Schneper, L.; Mathee, K. APMIS 2010, 118, 1-36.
- Papp-Wallace, K. M.; Endimiani, A.; Taracila, M. A.; Bonomo, R. A. Antimicrob. Agents Chemother. 2011, 55, 4943-4960.
- Galletti, P.; Giacomini, D. Curr. Med. Chem. 2011, 18, 4265-4283.
- Veinberg, G.; Potorocina, I.; Vorona, M. Curr. Med Chem. 2013, 21, 393-416.
- Kamath, A.; Ojima, I.; Tetrahedron 2012, 68, 10640-10664.
- Piens, N.; De Kimpe, N.; D’hooghe, M. Prog. Heterocycl. Chem. 2016, 28, 27-55.
- Dražić, T.; Roje, M. Chem. Heterocycl. Compd. 2017, 53, 953-962.
- a) Kiss, L.; Fülöp, F. Chem. Rec. 2018, 18, 266-281; b) Kiss, L.; Mándity, I. M.;· Fülöp, F. Amino Acids 2017, 49, 1441–1455.
- Zhou, Y.; Wang, Y.; Gu, Z.; Wang, S.; Zhu, W.; Acena, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 442-518.
- Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432-2506.
- O’Hagan D. J. Fluorine Chem. 2010, 131, 1071-1081.
- a) Kiss, L.; Forró, E.; Fustero, S.; Fülöp, F. Eur. J. Org. Chem. 2011, 4993-5001; b) Kiss, L.; Remete, A. M. Eur. J. Org. Chem. 2019, 5574–5602.
- Nonn, M.; Kiss, L.; Haukka, M.; Fustero, S.; Fülöp, F. A Org. Lett. 2015, 17, 1074-1077 (and references therein).
- Tarui, A.; Kondo, S.; Sato, K.; Omote, M.; Minami, H.; Miwa, Y.; Ando, A. Tetrahedron 2013, 69, 1559-1565.
- Sundell, R.; Siirola, E.; Kanerva, L. T. Eur. J. Org. Chem. 2014, 6753-6760.
- Trulli, L.; Raglione, V.; Fioravanti, S. Eur. J. Org. Chem. 2018, 3743-3749.
- Thi, H. D.; Thuy, G. L. N.; Catak, S.; Van Speybroeck, V.; Nguyen, T. V.; D’hooghe, M. Synthesis 2018, 50, 1439-1456.
- Thi, H. D.; Goossens, H.; Hertsen, D.; Otte, V.; Nguyen, T. V.; Van Speybroeck, V.; D’hooghe, M. Chem. Asian J. 2018, 13, 421-431.
- El Dine, A. N.; Grée, D.; Roisnel, T.; Caytan, E.; Hachem, A.; Grée, R. Eur. J. Org. Chem. 2016, 556-561.
- Kuznetsova, L.; Ungureanu, I. M.; Pepe, A.; Zanardi, I.; Wu, X.; Ojima, I. J. Fluorine Chem. 2004, 125, 487-500.
- Joyeau, R.; Felk, A.; Guillaume, S.; Wakselman, M.; Vergely, I.; Doucet, C.; Boggetto, N.; Reboud-Ravaux, M. J. Pharm. Pharmacol. 1996, 48, 1218-1230.
- Higman, C. S.; Lummiss, J. A. M.; Fogg, D. E. Angew. Chem. Int. Ed. 2016, 55, 3552-3565.
- Monsaert, S.; Vila, A. L.; Drozdzak, R.; Van Der Voort, P.; Verpoort, F. Chem. Soc. Rev. 2009, 38, 3360-3372.
- Prunet, J.; Eur. J. Org. Chem. 2011, 3634-3647.
- Smith, B. J.; Sulikowski, G. A. Angew. Chem. Int. Ed. 2010, 49, 1599-1602.
- Kardos, M.; Kiss, L.; Fülöp, F. Asian. J. Org. Chem. 2015, 4, 1155-1159.
- Kardos, M.; Kiss, L.; Haukka, M.; Fustero, S.; Fülöp, F. Eur. J. Org. Chem. 2017, 1894-1901.
- Fülöp, F.; Forró, E.; Tóth, G. K. Org. Lett. 2004, 6, 4239-4241.
- BouzBouz, S.; Cossy, J. Org. Lett. 2001, 3, 1451-1454.
- Boufroura, H.; Mauduit, M.; Drège, E.; Joseph, D. J. Org. Chem. 2013, 78, 2346-2354.
- Nonn, M.; Benke, Z.; Fustero, S.; Fülöp, F.; Kiss, L. Eur. J. Org. Chem. 2019, 5285–5293.
Статья рекомендована к публикации членом редколлегии Проф. József Rábai
Fluorine Notes, 2019, 127, 3-4


