Поступило в редакцию: Ноябрь 2019
Fluorine Notes, 2019, 127, 1-2
СИНТЕЗ 2-ФТОРОКТАНОВОЙ КИСЛОТЫ: ЭКСПЕРИМЕНТЫ В ЛАБОРАТОРИИ СОВРЕМЕННОЙ ОРГАНИЧЕСКОЙ ХИМИИ
Анико Немес (Anikó Nemes), * Денес Сабо (Dénes Szabó), Йожеф Рабай (József Rábai)
Институт химии Университета ELTE Eötvös Loránd, Будапешт 1117, Венгрия
E-mail: neagaft@caesar.elte.hu (Dr. Anikó Nemes)
Аннотация: Описана процедура многоступенчатого синтеза 2-фтороктановой кислоты (4), выполненная в рамках индивидуальных студенческих проектов в практикуме лаборатории современной органической химии (Advanced Organic Chemistry Laboratory). При этом в качестве теоретической основы студенты должны были изучить химию енолятов (реакцию Гелля-Фольгарда-Зелинского), а также реакции нуклеофильного замещения и элиминирования. В ходе практической работы ими были апробированы такие передовые лабораторные методики, как медленное введение реагентов, образование реагентов in situ и вакуумная дистилляция. После каждой стадии синтеза продукты реакции подвергались исследованию - определению температуры кипения/плавления и 1H, 13C, 19F ЯМР-спектроскопии.
Ключевые слова: α-фторалкановые кислоты, фторид калия, бромирование по реакции Гелля-Фольгарда-Зелинского, замещение, образование.
Теоретическое введение
Фторсодержащие органические соединения играют важную роль в материаловедении [1] и медицинской/фармацевтической химии [2], поэтому синтез этих соединений представляет большой интерес [3]. В последние несколько десятилетий около 20% новых активных фармацевтических веществ (API) содержали как минимум по одному атому фтора [4]. В процессе разработки лекарств осуществляют модификацию т. н. «ведущего» соединения для получения молекулы лекарственного препарата, обладающего оптимальными биологической активностью и фармакологическими свойствами. Замена атома водорода на атом фтора может влиять на связывание с белком-мишенью, на метаболизм молекулы в биологической системе и/или на фармакокинетические свойства препарата [5]. Введение атома фтора в ароматическое кольцо обычно оказывает влияние на связывание белка посредством π-π-взаимодействий, на сродство водородных связей и на окислительный метаболизм молекулы, тогда как алифатическое замещение фтора изменяет равновесную конформацию малой молекулы, тем самым влияя на связывание с белком [6]. По этим причинам синтез фторсодержащих молекул является важным навыком для химиков [7], [8], [9].
Несмотря на большую ценность фторорганических соединений, в большинстве учебников по органической химии отсутствует теория и методы синтеза, предназначенные для формирования C-F-связей. Несмотря на то, что ряд галоген-углеродных связей может формироваться относительно легко и избирательно, образование C-F-связей обычно происходит неселективно и требует использования агрессивных реагентов и специального оборудования. Простейшим способом синтеза фторсодержащих алифатических соединений является реакция замещения уходящей группы нуклеофильным фторидом (обычно в форме KF). Наиболее существенным недостатком подобных реакций является высокая степень побочной реакции элиминирования. Оптимальный выход продукта часто может достигаться за счет более низкой степени конверсии, поэтому этот продукт, помимо продуктов элиминирования, может содержать исходное вещество.
2-Фторкарбоновые кислоты можно синтезировать в три стадии, начиная с выбора подходящей карбоновой кислоты (см. схему на рисунке 1) для синтеза 2-фтороктановой кислоты 4. На первой стадии в одном сосуде происходят реакции галогенирования карбоновых кислот в α-положении (реакция Гелля-Фольгарда-Зелинского) и этерификация (т. е. превращение в сложный эфир), после чего атом брома образовавшегося метил 2-бромкарбоксилата (2) (см. схему 1) замещается фтором (с помощью KF в ацетамиде), и, наконец, сложноэфирная группа гидролизуется с получением 2-фторкарбоновой кислоты.
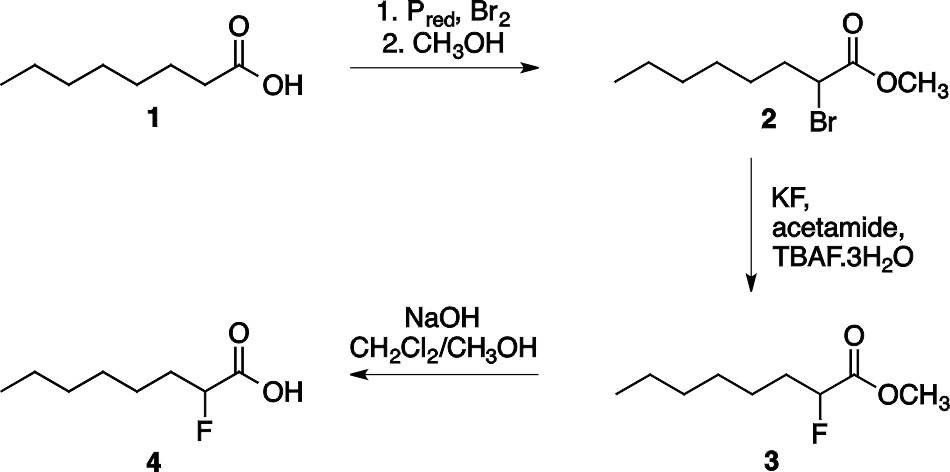
Схема 1. Синтез 2-фтороктановой кислоты 4.
Основной учебной целью выполнения данного индивидуального лабораторного задания являлась демонстрация сложности последовательностей линейного синтеза и важности введения теоретических основ химии в лабораторную практику. Вместе с тем, этот синтез позволяет знакомить студентов с передовыми лабораторными методиками (процедурами работ с высокоэкзотермическими и селективными реакциями, облегчающими разделение/очистку продуктов, вакуумной дистилляцей) и совершенствовать их лабораторные навыки. Эти эксперименты также были призваны подчеркнуть важность мониторинга отдельных реакций, а также определения структуры и соотношения реагентов методом ЯМР-спектроскопии.
Этот многоступенчатый синтез выполнялся студентами первого и второго года обучения в рамках курса Advanced Organic Chemistry Laboratory, где каждому студенту предлагались индивидуальные задания по синтезу.
Педагогические цели и результаты обучения
Синтез 2-фтороктановой кислоты был разработан с целью моделирования реальных научно-исследовательских работ. В лаборатории современной органической химии студенты получили оригинальные исследовательские задания по синтезу таких молекул, как молекулы 2-фторстеариновой кислоты [10], 2-фторгексановой кислоты [11] или других производных соединений с длинной цепью [12]. Они должны были адаптировать экспериментальные процедуры к исходным реагентам. Реакционная способность указанных соединений эквивалентна, однако их физические свойства, такие как растворимость, температуры плавления/кипения, консистенция и т. п.) различаются, поэтому студенты должны были модифицировать процедуры выделения и очистки этих соединений. Эти лабораторные задания по синтезу также можно выполнять и в «обычной» лаборатории органической химии, в которой преподаватель будет давать подробное экспериментальное описание реакций, начиная с реакции синтеза октановой кислоты. В любом случае студенты для определения соотношения продуктов реакции в качестве мощного аналитического метода исследований должны использовать метод ЯМР-спектроскопии.
Педагогические задачи
В процессе получения опыта лабораторного синтеза и применения на практике теоретических знаний о реакциях студенты знакомятся с методами тонкослойной хроматографии и ЯМР-спектроскопии.
- На первом этапе синтеза демонстрируется важность использования чистых исходных реагентов и работы с высокоэкзотермическими реакциями путем медленного введения одного из реагентов.
- На втором этапе синтеза демонстрируется важность знания механизмов реакции, включая потенциальные побочные реакции; использование различной химической реакционной способности побочного продукта, а также необходимость использования передовых аналитических методов.
- В процессе подготовки и оформления лабораторного отчета студенты могут закреплять свои знания о реакционной способности карбоновых кислот и их производных (а также об их относительной реакционной способности), а также об алифатических реакциях нуклеофильного замещения/элиминирования и базовой ЯМР-спектроскопии.
Важность обучения
В процессе обучения студенты могут:
- работать со стандартным лабораторным оборудованием
- использовать метод ЯМР-спектроскопии для определения качества и количества побочных продуктов, а также для определения их структуры
- эффективно по времени планировать последовательность синтеза
- работать с опасными веществами, инертными газами и вакуумным оборудованием
- оформлять подробные лабораторные отчеты, содержащие все необходимые сведения о физических свойствах исходных веществ/реагентов, степени их чистоты; об особенностях метода хроматографии и о расшифровке ЯМР-спектров).
После предварительного тестирования преподаватель знакомит студентов с существующими лабораторными методиками (с использованием такого лабораторного оборудования, как, например, реакторы, роторные вакуумные испарители, дистилляторы, вакуумные насосы), а также с теорией химических реакций. Для получения максимальных результатов обучения во время эксперимента преподаватель будет поддерживать индивидуальную устную обратную связь со студентами.
Краткое описание эксперимента
Трехстадийный синтез 2-фтороктановой кислоты (4) (см. схему 1) показан на схеме 1. Бромирование октановой кислоты (1) (см. схему 1) и её последующая этерификация позволяет получать метил 2-бромоктаноат (2) (см. схему 1) в качестве первого промежуточного соединения. На следующей стадии, помимо обмена галогеном, также происходит побочная реакция элиминирования с получением алкена (метил (Е) -2-октеноата (5) (см. схему 2). Этот побочный продукт не может быть отделен от целевых веществ методом дистилляции, поскольку их точки кипения достаточно близки (94-96 °C при 20 мм рт. ст. для продукта (3) и 89-91 °C при 9 мм рт. ст. для продукта (5) (см. схему 2). Для эффективного разделения эту реакционную смесь обрабатывают бромом, который превращает алкен в вицинальный дибромид (6) (см. схему 2), который обладает значительно более высокой температурой кипения, чем требуемый продукт, поэтому их можно было разделять методом вакуумной перегонки.
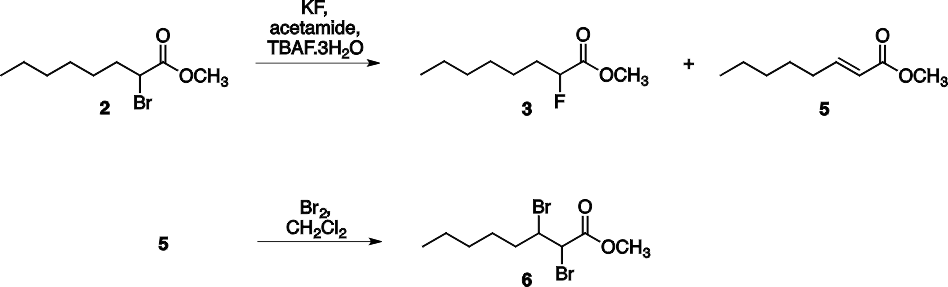
Схема 2. Реакции на второй стадии синтеза.
На третьей стадии протекает реакция гидролиза сложного эфира, что приводит к получению конечного продукта синтеза карбоновой кислоты (4) (см. схему 2). Студенты выполняли эксперименты индивидуально, с проведением 1Н и 13С ЯМР- после каждой стадии для определения структуры продукта (продуктов) реакции, а на второй стадии - для определения соотношения побочных продуктов реакций. Каждый эксперимент занимал примерно три недели, с восьмичасовым лабораторным периодом для его завершения.
Описание эксперимента
Синтез и очистку метил 2-бромоктаноата 2 (см. схему 2), а также подготовку к выполнению реакции замещения студенты проводили в течение 8 ч (для завершения этих операций большинству студентов обычно требовалось 7 ч). Исследование реакции замещения и очистку метил 2-фтороктаноата 3 (см. схему 2) они проводили в течение второго восьмичасового лабораторного периода (для завершения этих исследований большинству студентов обычно требовалось 6 ч). Гидролиз вышеуказанного сложного эфира, а также спектроскопические измерения и определение структуры студенты проводили в течение третьего восьмичасового лабораторного периода (для завершения этих операций большинству студентов обычно требовалось 8 ч). Письменные отчеты об этих процедурах студенты должны были представлять в течение 4 дней, а замечания и комментарии к этим отчетам выдавались в течение следующего лабораторного периода. Подробное описание этих экспериментов можно найти в разделе «Эксперимент».
Факторы опасности
Все эксперименты должны проводиться в хорошо вентилируемом вытяжном шкафу и с использованием соответствующих средств индивидуальной защиты. Студенты должны всегда надевать защитные очки и перчатки.
Все соединения, используемые в этих экспериментах, считаются раздражающими. Так, растворители вызывают раздражение кожи, глаз и дыхательных путей и, возможно, являются канцерогенными соединениями; кроме того, метанол в жидком и парообразном состоянии обладает высокой воспламеняемостью. Октановая кислота очень опасна при ее попадании в рот и желудочно-кишечный тракт, однако 2-фтороктановая кислота не обладает токсичностью [13,14]. NaOH и HCl являются агрессивными жидкостями. Жидкий элементарный бром или его пары могут вызывать повреждение тканей, особенно слизистых оболочек глаз, рта и дыхательных путей. Его контакт с кожей может вызывать ожоги, а вдыхание его паров - вызвать сильное раздражение дыхательных путей, которое может сопровождаться кашлем, удушьем или одышкой, и при серьезной передозировке - приводить к летальному исходу.
Полученные результаты и их обсуждение
Метод синтеза 2-фтороктановой кислоты из октановой кислоты был разработан и полностью протестирован авторами данной статьи, после чего полный процесс синтеза выполнялся 4 студентами-магистрами на протяжении двух семестров. Типичный выход этой кислоты у студентов в результате трехстадийного синтеза составлял 17% (на три стадии).
Первое бромирование может выполняться согласно общему методу. При этом важно подчеркнуть, что при использовании чистого красного фосфора реакция протекает намного быстрее, чем с фосфором технической чистоты. В первом случае метанол вводили в реакционную смесь через 2 ч, тогда как во втором случае реакция протекала только при дополнительном перемешивании этой смеси в течение 8 ч. Перед использованием красный фосфор обрабатывался кипятком, а затем фильтровался и просушивался. Чистый метил 2-бромоктаноат может быть синтезирован со средним выходом 89%.
Реакция замещения (2) (см. схему 2) протекала с использованием KF в качестве источника фтора в ацетамиде, в присутствии TBAF3H2O в качестве катализатора. Для подавления нежелательной побочной реакции и получения оптимального практического выхода продукта, реакцию проводили при температуре 75 °С в течение 60 ч, а реакционную смесь обрабатывали при достижении конверсии примерно 60 %.
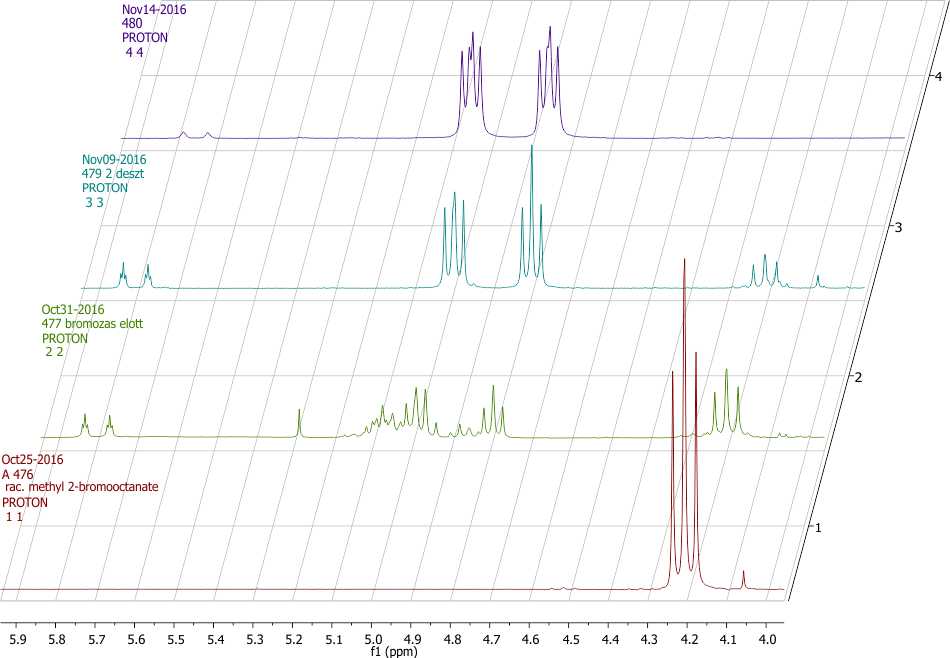
Схема 3. Частичные 1H ЯМР-спектры (4,0-6,0 м.д.) для соединений (2) (показан красным цветом, 11), для неочищенного соединения (3) (показан зеленым цветом, 22), для чистого соединения (3) (показан синим цветом, 33) и (4) (показан фиолетовым цветом, 44).
Соотношение продуктов реакции определялось методом 1H ЯМР-спектроскопии с использованием интегральных значений хорошо разрешаемых пиков CHCOOCH3 (см. схему 3). Пик метина (2) находится на спектре в виде дублета дублетов (dd) при 4,21 м.д., тогда как пик СН (3) – в виде дублета дублетов дублетов (ddd) при 4,90 м.д.. Алкеновый побочный продукт (5) содержит следы CHCOOCH3 при 5,78 и 5,82 м.д.. В стандартном эксперименте неочищенный продукт после реакции с KF содержал 46% замещенного продукта (3), с 14% 5 и 40% исходного вещества (2) (см. схемы 2 и 3).
Как уже указывалось выше, сырой продукт очищали в две стадии, т. е. сначала высушенный раствор CH2Cl2 неочищенного продукта (3) вступал в реакцию с элементарным бромом, а затем полученную смесь очищали методом фракционной перегонки в дистилляционной колонке Vigreaux при пониженном давлении. Типичный выход продукта у студентов на этом этапе составлял 36%. Как видно на схеме 3, этот продукт также содержал некоторое количество продуктов (2) (примерно 13%) и (5) (примерно 8%), однако было установлено, что при использовании этой смеси на последней стадии гидролиза продукт на выходе представлял собой чистую 2-фтороктановую кислоту (4). В другом варианте, метил 2-бромоктаноат 2 и метил 2-фтороктаноат 3 также можно разделять методом колоночной хроматографии, однако этот метод слишком времязатратен и требует большого объема растворителя.
Третьей и последней стадией в этой последовательности реакций был гидролиз сложного эфира (3) в смеси CH2Cl2 - CH3OH в основных условиях; типичный выход конечного продукта у студентов составлял примерно 53%.
Выводы
Рассмотренная процедура многоступенчатого синтеза помогла освоению студентами нескольких новых лабораторных методов, которые они ранее не выполняли самостоятельно (например, метода вакуумной дистилляции), а также применению метода ЯМР-спектроскопии для определения молекулярной структуры продуктов реакции и их соотношения. При этом студенты в процессе синтеза должны были применять широкий спектр своих теоретических знаний. Студенты также ознакомились с одной из методик, которую можно использовать в тех случаях, когда основной и побочный продукты обладают примерно одинаковой температурой кипения.
Экспериментальная часть
ЯМР-спектры (с изотопами 1H, 19F, 13C) регистрировались на спектрометре Bruker Avance (250 МГц) при комнатной температуре (295 ± 2 0К) в CDCl3. Химические сдвиги (δ) относительно внутренних стандартов приводятся в м.д.: TMS (δ = 0,00 для 1H) и CFCl3 (δ = 0,00 для 19F). Температуры плавления определялись с помощью прибора Boetius, предназначенного для измерения температуры микроплавления, и не корректировались. Тонкослойная хроматография проводилась с использованием пластин Merck SiO2; элюент – гексан CH2Cl2 (1: 1 по объему).
Синтез метил 2-бромоктаноата (2)
Суспензию из октановой кислоты (20,0 г, 138,5 ммоль) и красного фосфора (4,29 г, 138,5 ммоль) нагревали до 80 °С, а затем в нее добавляли по каплям бром (26,0 мл, 80,7 г, 505 ммоль) при постоянном перемешивании с такой скоростью, чтобы температура реакционной смеси оставалась ниже 90 °C (приблизительно в течение 2 ч). Суспензию при температуре 80 °C дополнительно перемешивали в течение 2 ч, затем охлаждали до 0 ° C и в течение 15 минут в нее добавляли 33 мл метанола. Реакционную смесь перемешивали в течение ночи при комнатной температуре, а затем кипятили в течение 3 ч. Смесь разделяли по фазам, нижнюю фазу промывали в 50 мл CH2Cl2. Объединенные органические фазы промывали в 50 мл воды и высушивали над Na2SO4. Раствор упаривали, а неочищенный продукт очищали методом перегонки в вакууме. Выход продукта в виде бесцветной жидкости с температурой кипения 122-124 °С при 20 мм рт. ст. составлял 28,9 г (88 %).
1H NMR (CDCl3) δ: 0.87 (3H, т, 3JHH = 6.7 Гц, CH3CH2), 1.2-1.5 (8H, м, CH3CH2CH2CH2CH2), 1.9-2.1 (2H, м, CH2CHBr), 3.76 (3H, с, COOCH3), 4.21 (1H, дд, 3JHH = 7.6 Hz, 3JHH = 7.2 Hz, CHBr).
13C NMR (CDCl3) δ: 14.4 (CH3CH2), 22.8 (CH2-7), 27.6 (CH2-4), 28.8 (CH2-5), 31.8 (CH2-6), 35.3 (CH2-3), 46.1 (CHBr), 53.2 (COOCH3), 170.8 (COOCH3).
Синтез метил 2-фтороктаноата (3)
Смесь метил 2-бромоктаноата (28,87 г, 122 ммоль), ацетамида (15 г), KF (12,7 г, 219 ммоль) и TBAF3H2O (2,06 г, 6,53 ммоль) перемешивали при 75 °C в течение 60 ч при температуре 60 °C в атмосфере азота. После охлаждения до комнатной температуры в реакционную смесь добавляли 180 мл воды и экстрагировали CH2Cl2 (3 × 70 мл). К этому раствору добавляли бром и постоянно перемешивали эту смесь до тех пор, пока не пропадал цвет брома. Объединенные органические фазы промывали раствором K2S2O5 (2 г) в 50 мл воды, насыщенным раствором NaHCO3 (2 × 100 мл) и насыщенным раствором NaCl (100 мл) и высушивали над Na2SO4. Растворитель удаляли в вакууме, а неочищенный продукт очищали методом перегонки с использованием дистилляционной колонки Vigreaux. Выход продукта в виде бесцветной жидкости с температурой кипения 94-96 °С при 20 мм рт. ст. составлял 10,2 г (47,4%)
1H NMR (CDCl3) δ: 0.87 (3H, т, 3JHH = 6.6 Hz, CH3CH2), 1.2-1.5 (8H, м, CH3CH2CH2CH2CH2), 1.7-2.0 (2H, м, CH2CHF), 3.78 (3H, с, COOCH3), 4.90 (1H, ддд, 3JHH = 5.9 Hz, 3JHH = 6.0 Hz, 3JHF = 49.2 Hz, CHF).
13C NMR (CDCl3) δ: 14.4 (CH3CH2), 22.9 (CH2-7), 24.7 (д, 3JCF = 3 Hz, CH2-4), 29.1 (CH2-5), 31.9 (CH2-6), 32.8 (д, 2JCF = 21 Hz, CH2-3), 52.6 (COOCH3), 89.4 (1JCF = 184 Hz, CHF), 170.9 (д, 2JCF = 24 Hz, COOCH3).
19F NMR (CDCl3) δ: -192.46.
Синтез 2-фтороктановой кислоты (4)
Раствор метил 2-фтороктаноата (8,00 г, 45 ммоль) в CH2Cl2 (80 мл) смешивали с раствором NaOH (5,45 г, 136 ммоль) в 60 мл метанола. Реакционную смесь перемешивали в течение ночи; затем растворитель удаляли в вакууме. Остаток растворяли в 170 мл воды, охлаждали до 0 0C и подкисляли с помощью 20 мл HCl. Осадок отфильтровывали и высушивали в вакууме над P2O5. Выход продукта в виде белого воскообразного твердого вещества с температурой плавления 51-53 °С составлял 3,93 г (54%). ( [19], температура плавления 51-53 °С).
1H NMR (CDCl3) δ: 0.87 (3H, т, 3JHH = 6.4 Hz, CH3CH2), 1.2-1.4 (8H, м, CH3CH2CH2CH2CH2), 1.7-1.9 (2H, м, CH2CHF), 4.95 (1H, ддд, 3JHH = 4.8 Hz, 3JHH = 6.7 Hz, 3JHF = 49.2 Hz, CHF), 13.10 (1H, уш. с., COOH).
13C NMR (CDCl3) δ: 14.2 (CH3CH2), 22.3 (CH2-7), 24.2 (d, 3JCF = 3 Hz, CH2-4), 28.5 (CH2-5), 31.4 (CH2-6), 32.1 (д, 2JCF = 21 Hz, CH2-3), 88.7 (1JCF = 180 Hz, CHF), 170.4 (d, 2JCF = 24 Hz, COOH).
Альтернативные методы
Альтернативные многостадийные методы синтеза α-фторкарбоксилатов связаны с раскрывающимися эпоксидами [15,16], с взаимодействием α-гидроксикислот с реагентом DAST [17] или с реагентом Ишикавы [18]; с взаимодействием алкенов с агентами [BrF], полученными в реакционной смеси из HF и источника электрофильного брома [19, 20]; или с созданием целевой α-фторной кислоты путем объединения двух фрагментов, один из которых уже содержит атом фтора [21, 22, 23], как это указывалось в работе Розена с сотр. [24]. Кроме того, более поздние исследователи сообщили об эффективном методе синтеза разветвленных и/или неразветвленных α-фторэфиров с помощью реакции электрофильного реагента AcOF-CH3CN, полученного in situ, с соответствующими кетенацеталями. Этот метод достаточно оперативен и также пригоден для получения короткоживущих изотопов 18F (t1/2 = 109,771 мин) меченых целевых соединений [24].
Некоторые из указанных в [19, 22, 23] путей синтеза позволяют удобно синтезировать получать рацемическую α-фтороктановую кислоту, поскольку такие F-содержащие строительные блоки, как диэтилмонофтормалонат {CHF(CO2C2H5)2} и этилфториодоацетат (CHIFCO2C2H5), недавно появились в продаже, однако нынешние цены на эти фторсодержащие прекурсоры могут стать ограничивающим фактором для внедрения этих новых методов в расширенный лабораторный практикум.
Благодарности
Эта работа выполнялась в рамках программы ELTE Institutional Excellence (1783-3 / 2018 / FEKUTSRAT), при поддержке Министерства человеческих ресурсов Венгрии.
Список литературы
- R. Berger, G. Resnati, P. Metrangolo, E. Weber, J. Hulliger, Organic Fluorine Compounds: A Great Opportunity for Enhanced Materials Properties, Chem. Soc. Rev., 2011, 40, 3496– 3508. DOI: 10.1039/C0CS00221F.
- J.-P. Begue, D. Bonnet-Delpon, Bioorganic and Medicinal Chemistry of Fluorine, John Wiley & Sons, Inc., 2008. DOI: 10.1002/9780470281895.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. delPozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok, H. Liu, Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011), Chem. Rev. 114 (2014) 2432- 2506. DOI: 10.1021/cr4002879.
- D. O’Hagan; Fluorine in Healthcare: Organofluorine Containing Blockbuster Drugs, J. Fluorine Chem. 131 (2010) 1071–1081. DOI: 10.1016/j.jfluchem.2010.03.003.
- I. Ojima (Ed.), Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell, Chichester, 2009. DOI: 10.1002/9781444312096.
- A. Postigo, Late-Stage Fluorination of Bioactive Molecules and Biologically-Relevant Substrates, Elsevier, 2019.
- L. M. Yagupolskii, Aromatic and Heterocyclic Compounds with Fluorine-Containing Substituents, Naukova Dumka, Kiev,1988.
- G. K. Surya Prakash, F. Wang, Fluorine: The New Kingpin of Drug Discovery, Chim. Oggi/ Chem. Today 30 (2012) 30-36.
- A. Harsanyi, G. Sandford, 2‑Fluoromalonate Esters: Fluoroaliphatic Building Blocks for the Life Sciences, Org. Process Res. Dev. 2014, 18, 981-992. DOI: 10.1021/op500141c
- S. A. Pogány, G. M. Zentner, C. D. Ringeisen, Aqueous Fluoride as a Nucleophile in the Synthesis of 2-Fluorostearic Acid. Synthesis, 1987, 718-719. DOI: 10.1055/s-1987-28058.
- P. Kalaritis, R. W. Regenye, Enantiomerically pure ethyl (R)- and (S)-2-fluorohexanoate by Enzyme-catalyzed Kinetic Resolution. Org. Synth., Coll. Vol. 8, p. 258 (1993). DOI: 10.1002/0471264180.os069.02.
- J. E. Oliver, R. M. Waters, W. R. Lusby, A Convenient Synthesis of α-Fluoro Carboxylic Acids. Synthesis, 1994, 273-275. DOI: 10.1055/s-1994-25457.
- H. Gershon, S. G. Schulman, A. D. Spevack, Organic Fluorine Compounds. III. Action of Perchloryl Fluoride on Substituted Ethyl Cyanoacetates and Animal Toxicities of the Fluorinated Products. J. Med. Chem., 1967, 10, 536-541. DOI: 10.1021/jm00316a008.
- B. C. Saunders, G. J. Stacey, Toxic Fluorine Compounds containing the C-F Link. Part I. Methyl Fluoroacetate and Related Compounds. J. Chem. Soc., 1948, 1773-1779. DOI: 10.1039/jr9480001773.
- H. Keul, B. Pfeffer, K. Griesbaum, Halogenepoxide, 7. Umlagerungsreaktionen bei der Umsetzung von substituierten 2‐Chlor‐ und 2,3‐Dichloroxiranen mit Silbertetrafluoroborat, Chem. Ber. 1984, 117, 2193-2204 https://doi.org/10.1002/cber.19841170613.
- A. O. Amanetoullah, M. M. Chaabouni, A. Baklouti, Synthesis of 2-Fluoro- Acids, Esters, and Amides From α-Dicyanoepoxides, Synth. Commun. 1996, 26, 1155-1161. https://doi.org/10.1080/00397919608003723.
- M. Schlosser, D. Michel, α-Fluoro analogues of inflammation inhibiting α-arylpropionic acids, Tetrahedron, 1996, 52, 8257-8262. https://doi.org/10.1016/0040-4020(96)00411-5.
- S. Watanabe, T. Fujita, M. Sakamoto, H. Endo, Fluorination of aromatic α-hydroxyesters with N,N-diethyl-1,1,2,3,3,3-hexafluoropropaneamine, J. Fluorine Chem. 1990, 47, 187-192. https://doi.org/10.1016/S0022-1139(00)82371-4.
- F. L. M. Pattison, R. L. Buchanan, F. H. Dean, The Synthesis of α-Monofluoroalkanoic Acids, Can. J. Chem. 1965, 43, 1700-1713. https://doi.org/10.1139/v65-224.
- O. Goj, S. Kotila, G. Haufe, Convenient routes to 2-aryl-2-fluoropropionic acids: Synthesis of monofluorinated analogues of (±)-ibuprofen, (±)-naproxen and related compounds, Tetrahedron 1996, 52, 12761-12774. https://doi.org/10.1016/0040- 4020(96)00758-2.
- A. Thenappan, D. J. Burton, Alkylation of (fluorocarbethoxymethylene)tri-n- butylphosphorane: a facile entry to α-fluoroalkanoates, J. Org. Chem. 1990, 55, 2311-2317. https://doi.org/10.1021/jo00295a016.
- Y. Wang, Z. Y. Yang, D. J. Burton, Nickel catalyzed reaction of iodofluoroacetates with alkenes as a facile route to α-fluoroesters, Tetrahedron Lett. 1992, 33, 2137-2140. https://doi.org/10.1016/0040-4039(92)88160-7.
- C. Zhi, Q.-Y. Chen, Novel and practical preparation of a-fluoro-functionalized esters from fluoroiodoacetates, J. Chem. Soc., Perkin Trans. 1, 1996, 1741-1747. DOI: 10.1039/P19960001741.
- S. Rozen, A. Hagooly, R. Harduf, Synthesis of α-Fluorocarboxylates from the Corresponding Acids Using Acetyl Hypofluorite, J. Org. Chem. 2001, 66, 7464-7468. https://doi.org/10.1021/jo010677k
Статья рекомендована к публикации членом редколлегии д.х.н. С.М. Игумновым
Fluorine Notes, 2019, 127, 1-2


