Поступило в редакцию: Июнь 2019
УДК 547.562.32
Fluorine Notes, 2019, 125, 1-2
Получение пентафторфенола и других полифторфенолов и полифтордигидроксибензолов из полифторароматических кислот
В.Э. Бойко, В.Л. Дон, С.М. Игумнов
Институт элементоорганических соединений им. А. Н. Несмеянова Российской академии наук, 119991, Москва,
ул. Вавилова, д. 28
e-mail: boykii@mail.ru
ЗАО НПО «ПиМ-Инвест», 119991, Москва, Ленинский проспект, 47
Аннотация: Разработан способ получения пентафторфенола и других полифторфенолов и полифтордигидроксибензолов из соответствующих ароматических кислот окислением цинкорганических соединений, получаемых декарбоксилированием триметилсилиловых эфиров этих кислот. Реакции получения цинкорганических соединений и их окисления проводят без выделения промежуточных продуктов.
Ключевые слова: пентафторфенол, пентафторбензойная кислота, окисление, полифторфенол, полифтордигидрокибензол, (триметил)силиловый эфир перфторбензойной кислоты.
Фторированные фенолы находят широкое применение для создания полимеров, обладающих уникальными свойствами [1], в качестве компонентов металлоценовых каталитических систем для полимеризации олефинов [2-4].
Образование пентафторфениловых эфиров широко используется в пептидном синтезе, поскольку пентафторфенол легко взаимодействуют с аминами с образованием амидных связей, таким образом пентафторфенол применяется в синтезе пептидов и нуклеозидов, являющихся интермедиатами в синтезе противоопухолевых агентов, ингибиторов ВИЧ [5-7].
Большинство известных методов получения пентафторфенола заключается в замещении фтора в гексафторбензоле на OH-группу взаимодействием его со щелочами, либо получении фенилалкиловых эфиров взаимодействием гексафторбензола с алкоголятами, и их последующем разложении с получением пентафторфенола.
Главным препятствием к промышленному использованию данных способов является то, что в настоящее время гексафторбензол не является промышленно доступным продуктом в связи с тем, что гексахлорбензол, из которого его получали ранее, запрещен к производству по причине его высокой канцерогенности. Именно поэтому возникает настоятельная потребность в способе синтеза пентафторфенола из промышленно доступного в настоящий момент сырья, а именно из пентафторбензойной кислоты, пентафторбензола или бромпентафторбензола.
Описаны получение пентафторфенола из бромпентафторбензола через пентафторфенилмагнийбромид, с последующим превращением его в пентафторфенилдиметоксиборат либо пентафторфенилборную кислоту взаимодействием с триметоксибором, и дальнейшим их окислением перекисью водорода [8-9], либо непосредственным окислением пентафторфенилмагнийбромида пероксидами [10]. Однако получение реактива Гриньяра в диэтиловом эфире или тетрагидрофуране является процессом повышенной опасности и требует использования инертной атмосферы.
Пентафторбензойная кислота и другие полифторароматические кислоты в настоящее время являются самыми доступными промышленными фторароматическими продуктами, поэтому целесообразно было искать путь к полифторфенолам исходя из полифторароматических кислот. Удобные методы получения пентафторфенил (триметил)силана из пентафторбензойной кислоты (декарбоксилированием солей ароматических кислот в присутствии силилирующих агентов или же декарбоксилированием соответствующих триметилсилиловых эфиров) были разработаны нами ранее [11,12]. Затем мы исследовали с целью получения пентафторфенола окисление триметил(пентафторфенил) силана и нашли, что при использования в качестве окислителя трет-бутилпероксибензоата в присутствии KF и CuCl в ДМФА окисление силана приводит к пентафторфенолу с выходом 65%, но реакция осложняется побочными процессами образования пентафторбензола и декафторбифенила. При использовании таких окислителей как перекись водорода и перекись трет-бутила пентафторбензол и вовсе является основным продуктом реакции, а образования пентафторфенола не происходит вовсе [13].
Дальнейшие поиски подходов к получению пентафторфенола и других полифторгидроксибензолов из соответствующих ароматических кислот привели к исследованию окисления полифторароматических цинк-органических соединений. Преимуществом цинк-органических соединений по сравнению с магний-органическими сооединениями является их большая стабильность, что позволяет работать с ними не в эфирных, а в полярных апротонных растворителях, таких как ДМФА.
Раствор цинк-органического соединения получали из пентафторбензойной кислоты в две стадии без выделения промежуточного продукта триметилсилилового эфира кислоты. Пентафторбензойную кислоту кипятили с избытком триметилхлорсилана, и полученный триметилсилиловый эфир без выделения затем взаимодействием с предварительно обезвоженными азеотропной отгонкой воды солями цинка или его окисью превращали в соответствующее цинковое производное.
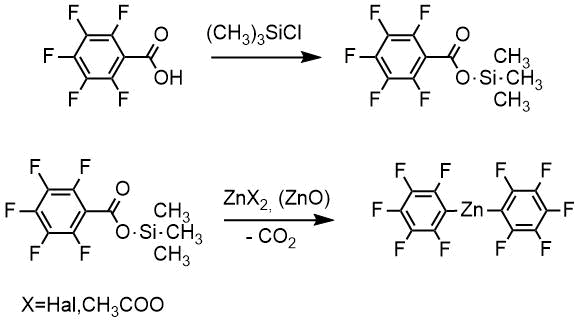
Полученный таким образом раствор цинкорганического соединения окисляли трет-бутилпероксибензоатом в присутствии солей одновалентной меди CuX, где X = Cl, Br, I (метод А), при этом получали трет-бутиловый эфир пентафторфенола, кипячением которого с соляной кислотой получали пентафторфенол. Суммарный выход на выделенный продукт 99% чистоты составил более 70%. метод А.
метод А
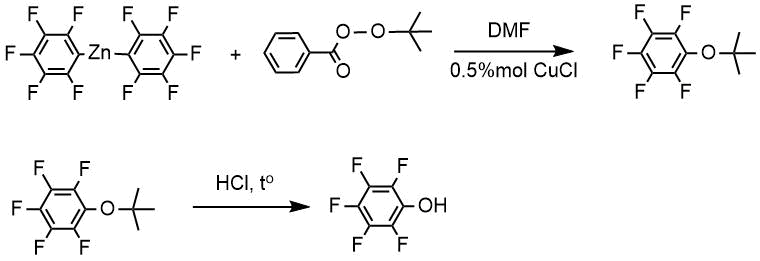
Если в качестве пероксисоедининения была взят комплекс перекиси водорода с мочевиной (метод В), то после проведения реакции ее с соответствующим цинковым производным и обработки реакционной смеси соляной кислотой сразу получали пентафторфенол, но выход пентафторфенола в этом случае был существенно меньше, поскольку наряду с пентафторфенолом получали пентафторбензол.
метод Б
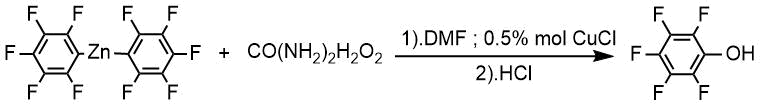
Из 2,4,5,6-тетрафторбензойной кислоты по методу А был получен 2,4,5,6-тетрафторфенол с выходом 76%.
Если в качестве исходного вещества взять дикарбоновую перфторароматическую кислоту, то в реакции с избытком триметилхлорсилана получают бис-триметилсилиловый эфир соответствующей кислоты и далее через промежуточное образование раствора бис-цинкового производного по методу А получают сначала бис-(трет-бутиловый) эфир, а затем соответствующий дигидрокситетрафторбензол.
Из тетрафтортерефталевой килоты в реакции с избытком триметилхлорсилана был получен бис-триметилсилиловый эфир тетрафтортерефталевой кислоты и далее по методу А тетрафторгидрохинон 99% чистоты с выходом 80%.
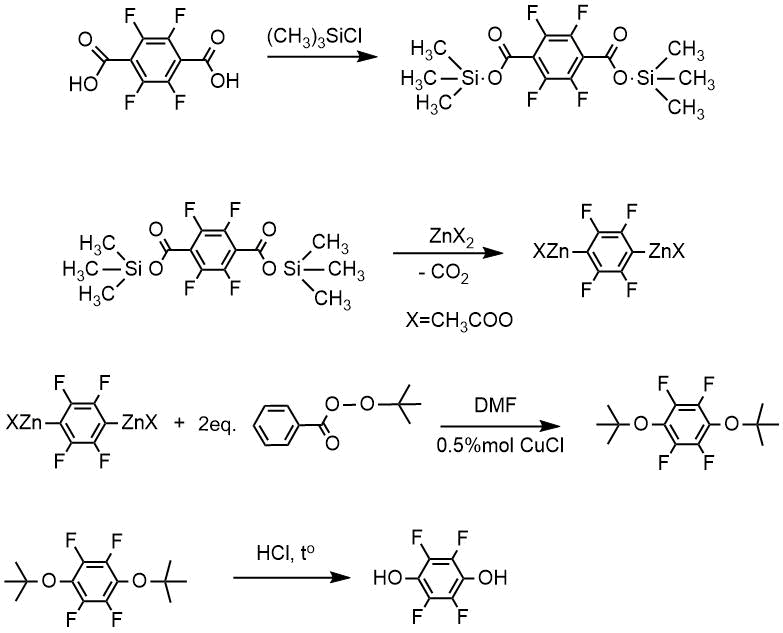
Таким образом, предложенный метод позволяет получать гидроксисоединения из соответствующиих ароматических кислот через промежуточное образование цинк-органических соединений без выделения промежуточных продуктов. На способ получения полифторгидроксибензолов получен патент РФ No 2536872.
Экспериментальная часть
ЯМР 1H, 19F спектры записаны на спектрометре “Bruker AVANCE-300” при 300 и 282 MHz, соответственно, внешний стандарт CDCl3. Химические сдвиги сигналов 1H спектров определены относительно остаточного сигнала растворителя (CHCl3 δ: 7,26) и даются в м.д. относительно ТМС. Химические сдвиги сигналов в спектрах 19F приведены в м.д. относительно CFCl3. Слабопольные сдвиги имеют положительное значение.
Триметилсилиловый эфир пентафторбензойной кислоты
К 2300 г (21,18 моль) триметилхлорсилана при перемешивании порциями присыпают 1493 г (7,04 моль) пентафторбензойной кислоты. Реакционную смесь нагревают до кипения и кипятят до завершения газовыделения. Затем отгоняют избыток триметилхлорсилана до температуры в кубе 120-130°С и получают 2000 г триметилсилилового эфира пентафторбензойной кислоты, который используют в следующей стадии без очистки. Выход количественный.
Спектр 19F ЯМР δ, м.д.:-140 м.(2F), -152 м.(1F), -163 м. (2F). 1H ЯМР δ, м.д.: 0,78c.
Трет-бутоксипентафторбензол
Смесь 800 г толуола, 1200 г ДМФА и 430 г (1,51 моль) ZnAc22H2O нагревают до кипения и отгоняют воду с насадкой Дина-Старка. После прекращения выделения воды насадку Дина-Старка и обратный холодильник заменяют соответственно капельной воронкой и системой для отгонки, состоящей из дефлегматора, холодильника Либиха, алонжа и приемной колбы, и к реакционной смеси по каплям добавляют 1000 г (3,52 моль) триметилсилилового эфира пентафторбензойной кислоты, после чего нагревают куб до 140-150°С, отгоняя фракцию с т.кип. 120-130°С. При перемешивании добавляют сначала 2 г (0,02 моль) CuCl, а затем при температуре 20-25°С по каплям добавляют 760 г (3,91 моль) трет-бутилпероксибензоата. Реакционную смесь перемешивают в течение 4 часов, охлаждают до температуры 10-15°С, добавляют 2 л 10%-ной соляной кислоты, перемешивают еще 20 минут. Верхний слой декантируют, к нижнему добавляют 2-2,5 объема воды и перегоняют с паром.
Получают 783 г сырца, содержащего по данным ГХ 88% трет-бутокси-пентафторбензола, который используют далее без дополнительной очистки.
Спектр 19F ЯМР δ, м.д.: -153,5 м.(2F), -165,0 м.(1F), -166,8 м. (2F). 1H ЯМР δ, м.д.: 0,78c.
Пентафторфенол. (метод A)
К 783 г сырца трет-бутоксипентафторбензола добавляют двухкратный объем 35% соляной кислоты, нагревают до кипения и кипятят в течение 4 часов (до окончания газовыделения). Затем реакционную массу охлаждают до 10°С, водный слой декантируют, а органический (нижний) ректифицируют и выделяют 475 г пентафторфенола чистотой 99% (по ГХ), т.кип. 139-142°С, т.пл. 34-36°С. Выход 72%.
Спектр 19F ЯМР δ, м.д.: -165,5 м.(2F), -168,6м.(2F), -174,6 м. (1F).
Пентафторфенол. (метод B)
Смесь 800 г толуола, 1200 г ДМФА и 122 г ZnO (1,51 моль) нагревают до кипения и кипятят с насадкой Дина-Старка, отделяя воду. После прекращения выделения воды насадку Дина-Старка и обратный холодильник заменяют соответственно капельной воронкой и системой для отгонки, состоящей из дефлегматора, холодильника Либиха, алонжа и приемной колбы, и к реакционной смеси по каплям добавляют 1000 г (3,52 моль) триметилсилилового эфира пентафторбензойной кислоты, после чего куб нагревают до температуры 140-150°С, отгоняя фракцию с т.кип. 120-130°С. Реакционную смесь охлаждают до 0°С, добавляют 2 г хлористой меди и затем порциями добавляют 500 г гидроперита и перемешивают еще 1-2 часа Затем содержимое колбы при перемешивании выливают в 2,5 л 10% раствора соляной кислоты, перемешивают еще 10 минут, отделяют нижний слой и перегоняют его с паром над концентрированной соляной кислотой. Полученный сырец ректифицируют и получают 360 г пентафторфенола чистотой 99% и 267 г предгона, содержащего пентафторбензол. Выход составляет 56%.
Триметилсилиловый эфир 2,3,5,6-тетрафторбензойной кислоты
К 202 г (1,86 моль) триметилхлорсилана при перемешивании присыпают порциями 120 г (0,62 моль) 2,3,5,6-тетрафторбензойной кислоты. Реакционную смесь нагревают до кипения, до завершения газовыделения, а затем отгоняют избыточный триметилхлорсилан до 120-130°С в кубе. Получают 165 г триметилсилилового эфира 2,3,5,6-тетрафторбензойной кислоты, который используют далее без дополнительной очистки.
19F ЯМР(CDCl3), δ, м.д.:-139,8 м. (2F), -140,6 м. (2F), 1H ЯМР(CDCl3) δ, м.д.: 0,78c.
2,3,5,6-тетрафторфенол (метод A)
В колбу помещают 200 г толуола и 68 г дигидрата ацетата цинка, реакционную смесь нагревают до кипения (110-115°С) и отгоняют воду. После прекращения выделения воды реакционную смесь остужают, воду отделяют взвешивают, если все необходимое количество воды выделилось, то к реакционной смеси добавляют 300 г ДМФА и снова нагревают до кипения, отгоняя толуол до температуры 130°С в парах. Реакционную смесь охлаждают и при перемешивании из капельной воронки добавляют 165 г (0,62 моль) триметилсилилового эфира 2,3,5,6-тетрафторбензойной кислоты. При этом наблюдается разогрев реакционной смеси и растворение ацетата цинка. Затем реакционную смесь нагревают, при температуре 80-85°С начинается газовыделение и в приемник начинает скапывать ацетат триметилсилана. Отгонку продолжают до температуры куба 140-150°С.
К полученному раствору цинкового производного 2,4,5,6-тетрафторбензола при перемешивании добавляют 0.6 г CuCl и при температуре 20-25°С по каплям добавляют 120.4 г (0,62 моль) трет-бутилпероксибензоата. По завершении добавления реакционную смесь перемешивают в течении часа. Затем содержимое колбы выливают в перемешивающийся раствор 0,5 л 10% соляной кислоты, перемешивают 10 минут, отделяют нижний слой, который далее кипятят с концентрированной соляной кислотой в течение 2 часов, после чего нижний слой отделяют и перегоняют с паром над концентрированной соляной кислотой. Получают 78 г 2,4,5,6-тетрафторфенола чистотой 99%. (76% выход).
19F ЯМР(CDCl3), δ, м.д.:-141,7м. (2F), -161,7м. (2F).
Бис-(триметилсилиловый) эфир тетрафтортерефталевой кислоты
К 50 г (0,21 моль) тетрафтортерефталевой кислоты добавляют 182 г (1,69 моль) триметилхлорсилана, кипятят до прекращения газовыделения, отгоняют избыток триметилхлорсилана. Получают 80,2 г (0,21 моль) бис-(триметилсилилового)эфира тетрафтортерефталевой кислоты.
19F ЯМР(CDCl3), δ, м.д.:-141м (4F), 1H ЯМР(CDCl3) δ, м.д.: 0,78c.
Тетрафторгидрохинон
К 80,2 г (0,21 моль) бис-(триметилсилилового)эфира тетрафтортерефталевой кислоты добавляют 250 мл ДМФА и 38,5 г безводного ацетата цинка и нагревают реакционную смесь с одновременной отгонкой триметилсилилацетата и декарбоксилированием. После окончания газовыделения, реакционную смесь охлаждают до 0°С, присыпают 0,2 г однохлористой меди и по каплям добавляют 81 г (0,42 моль) трет-бутилпероксибензоата. После окончания добавления реакционную смесь перемешивают дополнительно 2 часа при 0°С и затем 6 часов при комнатной температуре, после чего выливают на лед с 10% соляной кислотой и отделяют нижний слой. К нижнему слою добавляют двухкратный объем соляной кислоты, кипятят 3 часа, охлаждают, декантируют водный слой, нижний слой перегоняют и получают 30 г тетрафторгидрохинона чистотой 99%. Выход 80%.
19F ЯМР(CDCl3), δ, м.д.:-165,3м (4F).
Благодарности
Работа выполнена при поддержке Министерства науки и высшего образования Российской Федерации с использованием научного оборудования Центра исследования строения молекул ИНЭОС РАН.
Список литературы
- B. Boutevin,’ A. Rousseau,’ D. Bosc J. Polym. Sci. A, Polym.Chem., 1992, vоl. 30(7), p.1279–1286.
- Патент США № 8501655, 2013.
- Franz A.R. Kaul, Gerd T. Puchta, Horst Schneider, Manja Grosche, Dimitrios Mihalios, Wolfgang A. Herrmann J. Organomet. Chem., 2001, vol. 621, p.184.
- Metz, Matthew V.; Sun, Yimin; Stern, Charlotte L.; Marks, Tobin J. Organometallics, 2002, 21(18), p.3691-3702.
- Патент США № 7262169, 2007.
- WO 9616931, 1996.
- Jérôme Grugier, Juan Xie, Isabelle Duarte, Jean-Marc Valéry J. Org. Chem., 2000, vol. 65(4), p. 979.
- Korenaga T., Kobayashi F., Nomura K., Nagao S., Sakai T. J. Fluorine Chem., 128, 2007, p. 1153-1157.
- Заявка на патент Японии JP 82548(A), 2005.
- Заявка на патент Китая CN1847210 (A), 2006.
- Патент РФ№2507209.
- Патент РФ №2521168.
- В.Э. Бойко, A.A. Тютюнов, В.Л. Дон, С.М. Игумнов, Fluorine Notes, 2013, 91, c. 9-10.
Статья рекомендована к публикации членом редколлегии к.х.н. А.А. Тютюновым
Fluorine Notes, 2019, 125, 1-2


