Поступило в редакцию: Декабрь 2018
УДК 547.361
Fluorine Notes, 2018, 121, 9-10
1,1-Дихлоргексафторизобутилен и 2,2-дихлор-3,3-бис(трифторметил)оксиран – малотоксичные синтетические эквиваленты перфторизобутилена и его производных
A.A. Тютюновab, А.В. Синькоab, Н.Д. Каграмановa, С.Р. Стерлинa, С.М. Игумновab
aФедеральное государственное бюджетное учреждение науки Институт элементоорганических соединений им. А.Н. Несмеянова Российской академии наук, 119991, ГСП-1, Москва, В-334, ул. Вавилова, д. 28
bЗАО НПО “ПиМ-Инвест”, 119991, Москва, ул. Вавилова, д. 28
e-mail: tuytuynov@rambler.ru
Аннотация: На основе 1,1-дихлоргексафторизобутилена и 2,2-дихлор-3,3-бис(трифторметил)оксирана разработаны методы синтеза производных α-галоген- и α-гидрогексафторизомасляных кислот и алифатических соединений, содержащих полифторованные трет-бутильные группы.
Ключевые слова: 1,1,1-трихлор-3,3,3-трифтор-2-(трифторметил)пропан-2-ол; 1,1-дихлоргексафторизобутилен; 2,2-дихлор-3,3-бис(трифторметил)оксиран; 2-хлор-2-фтор-3,3-бис(трифторметил)оксиран.
Перфторизобутилен (ПФИБ), являясь одним из наиболее электрофильных фторолефинов, чрезвычайно легко взаимодействует с нуклеофильными реагентами с образованием продуктов присоединения или замещения [1-2]. В частности, при действии KF или CsF ПФИБ легко образует перфтор-трет-бутильный анион, реакции которого подробно рассмотрены в обзорах [3-4]. Следует добавить, что перфторизобутилен является исходным соединением для получения таких продуктов как бис(трифторметил)кетен, тиокетен [5-7] и производных перфторметакриловой кислоты [8], представляющих несомненный синтетический интерес.
Основным источником ПФИБ является фракция фторуглеродов С-4, получающаяся при синтезе тетрафторэтилена и гексафторпропилена пиролизом фреона 22. Однако, усовершенствование технологии пиролиза – проведение процесса в присутствии водяного пара – позволяет практически полностью исключить образование фракции С-4 (ПФИБ, перфторбутен-2 и перфторциклобутан) [9]. Это важное с точки зрения техники безопасности изменение технологии – перфторизобутилен чрезвычайно токсичен – одновременно ставит задачу изыскания безопасных, препаративных методов синтеза соединений, которые получают на основе ПФИБ.
Ранее было показано, что 1,1-дихлоргексафторизобутилен (1) в ряде случаев реагирует аналогично ПФИБ, а также является существенно менее токсичным соединением, чем перфторизобутилен (LCt50>25000 и 880 мг.мин.м-3 соответственно) [10]. Поэтому на практике значительно удобней и безопасней использовать вместо газообразного ПФИБ (т.кип. 6°C) его достаточно высококипящий и относительно безопасный аналог 1 (т.кип. 74°C) [11].
Целью данной работы является разработка методов синтеза соединений, содержащих перфтор-трет-бутильную группу, а также производных гексафторизомасляной кислоты без использования перфторизобутилена. Базовым соединением такого исследования мог бы явиться 1,1,1-трихлор-3,3,3-трифтор-2-(трифторметил)пропан-2-ол (2) – соединение, содержащее пергалоидированную трет-бутильную группу, простые методы синтеза которого из гексафторацетона и трихлоруксусной кислоты или трихлорацетата натрия разработаны ранее [12-13].
Известно, что третичный спирт 2 под действием PCl5 дезоксихлорируется, давая 1,1-дихлоргексафторизобутилен (1), однако выход 1 после 8-ми часового кипячения реакционной смеси не превышает 42% (при 40% конверсии 2) [14]. Принимая во внимание тот факт, что вицинальные галогенгидрины гладко восстанавливаются в олефины цинком в среде спирта или уксусной кислоты [15-16], нами исследована возможность восстановления 2 в 1, как альтернативного пути синтеза дихлоризобутилена 1.
Оказалось, что взаимодействие 2 с цинковой пылью в уксусной кислоте или сульфолане приводит к восстановлению трихлорметильной группы и образованию смеси соответствующих третичных спиртов. Причем в случае осуществления этой реакции в воде в присутствии хлористого аммония удается селективно синтезировать дихлоргексафтор-трет-бутанол 3.
Схема 1
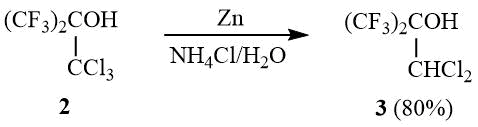
Трансформация ОН-группы в 2 в сложноэфирную-группу с повышенной нуклеофугностью, например CH3CO2, CF3CO2, ClCH2CFClCO2 или CH3SO3, позволяет превращать эфиры 4a-c реакцией с цинком в дихлоризобутилен 1 с препаративным выходом (60-80%):
Схема 2
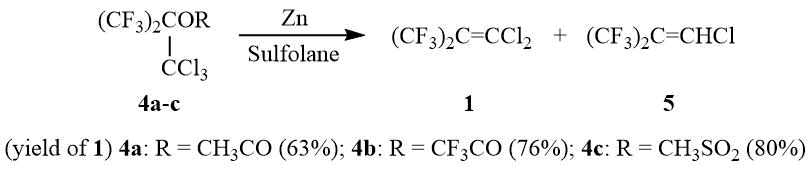
В отличие от ПФИБ образование перфтор-трет-бутильного аниона 6 из дихлоризобутилена 1 под действием KF в таких растворителях как диглим, ацетонитрил, DMF не наблюдается даже при нагревании или в присутствии 18-краун-6, Ph4PBr, (Et2N)3CCl. Также дихлоризобутилен 1 не образует 6 с CsF в диглиме. Однако действие трех эквивалентов CsF на 1 в ацетонитриле или DMF приводит к образованию 6 (ЯМР 19F δ: -47 (уш.с, (CF3)3С-)), который далее может использоваться в реакциях с различными электрофильными реагентами для синтеза соединений, содержащих перфтор-трет-бутильную группу, например 7a-e, а также мономера 8.
Схема 3
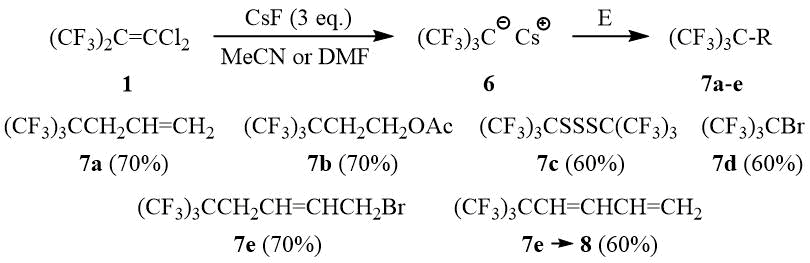
В тоже время реакция олефина 1 с МеОН в присутствии KF или KOH приводит к образованию хлоргексафторизобутенилметилового эфира 9 в качестве основного продукта реакции в результате нуклеофильного присоединения метанола с последующим дегидрохлорированием. Ранее аналогичной реакцией 1 с арилтиолами были получены арилхлоргексафторизобутенилсульфиды [11].
Схема 4
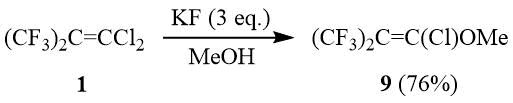
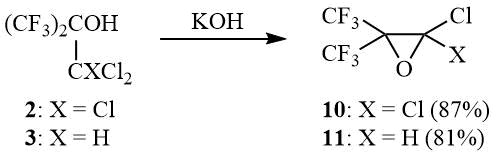
Другим важным аспектом химии вицинальных галогенспиртов, к которым можно отнести 1,1,1-трихлор-3,3,3-трифтор-2-(трифторметил)пропан-2-ол (2), является их способность подвергаться под действием оснований дегидрогалогенированию с образованием эпоксидов, в том числе фторсодержащих [17-18].
Так, ранее было показано, что 2 дегидрохлорируется водной щелочью с образованием окиси 1,1-дихлоргексафторизобутилена (10) с 50%-ным выходом [12]. В ходе работы было установлено, что реакция 2 с твердым КОН позволяет повысит выход окиси 10 до 87%. Аналогично трет-бутанол 3 был превращен в окись 11 с выходом 81%:
Схема 5
В то же время хлороктафтор-трет-бутанол (CF3)2C(OH)CF2Cl (12) не дегидрохлорируется KOH даже при длительном кипячении.
Высокая основность фторид-иона [19-20] давала основание полагать, что реакция трет-бутанола 2 c KF в апротонной среде также будет приводить к образованию окиси 10. Действительно спирт 2 реагирует с KF в диглиме при 100÷140°С, однако в качестве основного продукта реакции был получен перфторизобутироилфторид (13) с выходом ~50%.
Схема 6
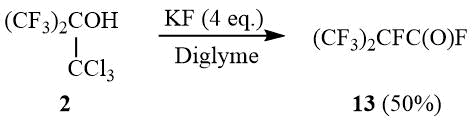
Очевидно, ацилфторид 13 образуется в результате реакции Йоцича-Рива [21-22], интермедиатом которой является гем-дихлорэпоксид 10, раскрывающийся фторид-анионом c образованием перфторизобутироилхлорида и последующим фторированием хлорформильной группы.
Дальнейшее исследование реакции окиси 10 с фтористым калием показало, что в таких апротонных растворителях как диглим, ацетонитрил или сульфолан в присутствии KF оксиран 10 изомеризуется с образованием хлорангидрида α-хлорперфторизомасляной кислоты (14).
Схема 7
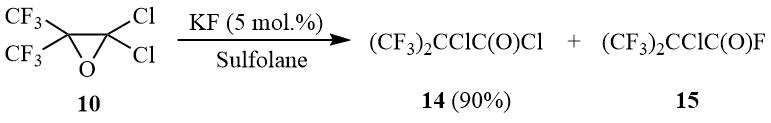
При увеличении количества вводимого в реакцию KF образуется α-хлорперфторизобутироилфторид (15) в качестве основного продукта реакции. Изобутироилгалогениды 14-15 являются удобными исходными соединениями для синтеза бис(трифторметил)кетена [23].
В свою очередь при взаимодействии окиси 10 с избытком фтористого калия в метаноле наблюдается образование исключительно метилового эфира перфторизомасляной кислоты (16).
Схема 8
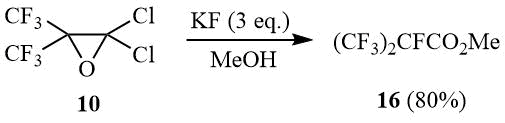
Очевидно, что образование α-галогенгексафторизобутироилгалогенидов 13-15 является следствием нуклеофильной атаки окиси 10 по третичному атому углерода галогенид-анионом (аналогично раскрытию окиси перфторизобутилена [24-25]). В том случае, когда в растворе присутствует каталитическое количество KF, например в таких растворителях как диглим или сульфолан, окись 10 преимущественно изомеризуется в хлорангидрид α-хлорперфторизомасляной кислоты (14). В растворе метанола, в котором растворимость KF достаточно высока, наблюдается раскрытие окиси 10 фторид-анионом и образование 16.
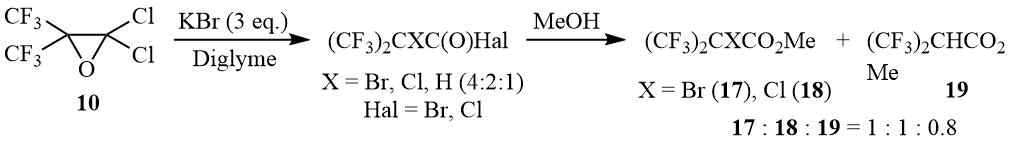
Под действием избытка KBr в диглиме окись 10 также раскрывается с образованием галогенангидридов α-бром-, α-хлор- и α-гидрогексафторизомасляных кислот в мольном соотношении 4:2:1. Очевидно, что образование α-хлорперфторизобутироилгалогенидов является следствием конкурентной атаки оксирана 10 хлорид-анионом, тогда как галогенангидриды -гидроперфторизомасляной кислоты по всей вероятности образуются в результате галогенофильной атаки третичного атома брома. Данное предположение подтверждается появлением брома при этерификации реакционной массы метанолом и резким изменением относительного содержания α-бром- и α-гидрогексафторизобутиратов (мольное соотношение эфиров 17:18:19 = 1:1:0,8), что скорее всего связано с большей растворимостью галогенидов калия в смеси МеОН/диглим.
Схема 9
Интересно отметить, что при взаимодействии 10 с избытком триэтиламина в метаноле с последующим подкислением реакционной массы был получен 2-хлор-1,1,1,3,3,3-гексафторпропан (20), образование которого можно объяснить декарбоксилированием промежуточно образующейся α-хлоргексафторизомаслянной кислоты.
Схема 10
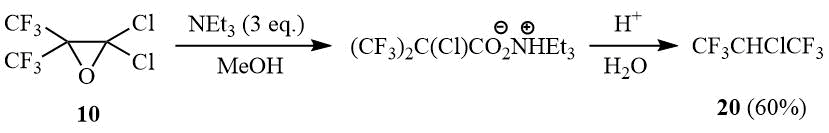
Ранее было показано, что окись перфторизобутилена под действием SbF5 изомеризуется в перфторизобутироилфторид [26]; причем та же реакция, но в присутствии HF, приводит к образованию перфтор-трет-бутанола [27]. В свою очередь карбинол 2 может быть превращен по реакции Свартса в дихлорфтор-, хлордифтор- и перфтор-трет-бутанол [12, 28-29]. Нам представлялось интересным исследовать взамодействие окиси 10 с пятифтористой сурьмой.
Как оказалось реакция оксирана 10 с SbF5 (0,5 эквивалента) при 20÷25°С приводит к образованию вязкой реакционной массы, которая, после перемешивания в течение 1 часа и разложения водой, дает смесь продуктов реакции, содержащей 25% оксирана 21 и 63% хлороктафтор-трет-бутанола (12). Получение карбинола 12 позволяет предположить, что его интермедиатом является (CF3)2C(OSbF3Cl)CF2Cl. В то же время нагревание реакционной смеси с тем же соотношением реагентов с одновременной отгонкой летучих продуктов реакции приводит к образованию оксирана 21 с 80%-ным выходом:
Схема 11
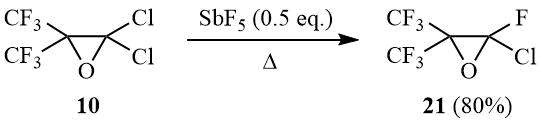
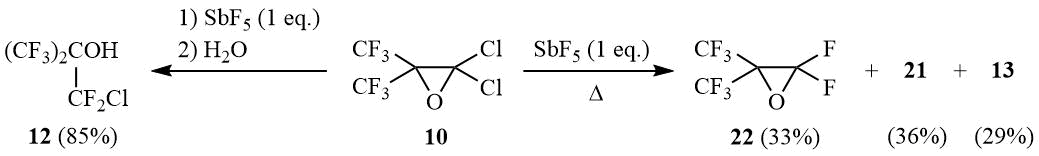
Взаимодействием эквимольных количеств оксирана 10 и SbF5 в аналогичных условиях (20÷25°С, часовое перемешивание с последующим разложением реакционной смеси водой) был получен карбинол 12 с 85%-ным выходом. Постепенное нагревание этой же реакционной смеси до 180÷190°С с одновременной отгонкой летучих продуктов реакции приводит к образованию смеси примерно равных количеств окиси перфторизобутилена (22), окиси 21 и перфторизобутироилфторида 13:
Схема 12
Таким образом, показано, что дихлоргексафторизобутилен 1 и его окись 10 представляют несомненный интерес в качестве исходных веществ для получения соединений, содержащих полифторированные изопропильные или трет-бутильные группы. В частности, дихлоргексафторизобутилен 1, являясь синтетическим эквивалентом высокотоксичного перфторизобутилена, может использоваться для введения перфор-трет-бутильной группы в различные классы органических соединений, а на основе окиси 10 могут быть получены производные α-галоген- и α-гидрогексафторизомасляных кислот.
Экспериментальная часть
ЯМР 1H, 19F спектры записаны на спектрометре “Bruker AVANCE-300” при 300 и 282 MHz, соответственно, внешний стандарт CDCl3. Химические сдвиги для 1H спектров приведены относительно остаточного сигнала растворителя (CHCl3 δ: 7,26) и даются в м.д. относительно ТМС. Химические сдвиги спектров 19F приведены в м.д. относительно CFCl3. Слабопольные сдвиги имеют положительное значение. Масс-спектры записаны на масс-спектрометре Finnigan Polaris Q (Trace GC ultra). Элементный анализ выполнен в Лаборатории микроанализа ИНЭОС РАН.
1,1,1-Трихлор-3,3,3-трифтор-2-(трифторметил)пропан-2-ол (2).
Получен по описанному ранее методу из гексафторацетона и CCl3CO2Na [13] или с использованием CCl3CO2SiMe3 по методу представленному ниже:
В 6-ти литровую колбу, снабженную механической мешалкой, термометром и обратным холодильником (-78°С), соединенного со склянкой Тищенко с конц. H2SO4, вносят 3,5 л диметилформамида и при перемешивании прибавляют 500 г (8,6 моль) сухого фтористого калия. В полученную суспензию при 15÷20°С вводят 1000 г (6 моль) гексафторацетона, смесь охлаждают до 5÷10°С и прибавляют по каплям 1550 г (6,58 моль) триметилсилилтрихлорацетата. Затем реакционную массу отогревают до ~25°С, перемешивают 4-5 часов, оставляют на ночь, постепенно, при 15÷20°С, прибавляют 516 г (2,19 моль) триметилсилилтрихлорацетата, смесь перемешивают 4-5 часов при ~25°С, оставляют на ночь. Полноту реакции контролируют по данным ЯМР 19F: в спектре должен наблюдаться сигнал 2 (-70 м.д.) и отсутствовать сигнал ГФА (-83 м.д.).
Реакционную смесь промывают 6-8 л одномолярной HCl-к-той, органический слой отделяют, промываю два раза равным объемом 5% соляной кислоты, смешивают при 15÷20°С с половинным объемом концентрированной серной кислоты, нагревают до 60÷70°С, отгоняя триметилфторсилан, остаток перегоняют в вакууме, собирая фракцию с т.кип. 50÷70°С/20 Торр.
Повторной перегонкой получают 1200 г (70%) карбинола 2, т.кип. 30°С/10 Торр (лит. 136÷137°С [28-29]). ЯМР 1H δ: 3,6 (c, OH); ЯМР 19F δ: -70,5 (с, CF3).
2-(Дихлорметил)-1,1,1,3,3,3-гексафторпропан-2-ол (3).
К смеси 151 г (0,53 моль) 2 и раствора 140 г (2,6 моль) хлористого аммония в 500 мл воды при интенсивном перемешивании и температуре 20÷25°С прибавляют 36 г (0,55 моль) Zn-пыли порциями по 6 г с интервалом в ~1-5 минут. Затем реакционную смесь перемешивают 1-1,5 часа, подкисляют, нагревают до четкого разделения фаз, нижний слой отделяют и перегоняют из равного объема концентрированной серной кислоты.
Ректификацией полученного дистиллята получают 106 г (80%) карбинола 3, т.кип. 114÷115°С (лит. 115°С [29]). ЯМР 1H: δ 3,5 (уш.c, 1H, OH), 5,7 (c, 1H, CHCl2); ЯМР 19F: δ -73,7 (с, CF3).
1,1,1-Трихлор-3,3,3-трифтор-2-(трифторметил)пропилацетат (4a).
К раствору 142,7 г (0,5 моль) карбинола 2 в 500 мл ацетонитрила при перемешивании и температуре 5÷10°С прибавляют 53,1 г (0,525 моль) триэтиламина и добавляют по каплям 41,2 г (0,525 моль) ацетилхлорида. Смесь перемешивают 3 часа при температуре 20÷25°С и выливают в двукратный объем холодной воды, нижний слой отделяют, промывают два раза двукратным объемом холодной воды, добавляют 10 г P2O5 и перегоняют в вакууме (10-15 Торр) собирая фракцию, выкипающую при 55÷65°С.
Повторной перегонкой получают 142 г (87%) эфира 4a, т.кип. 62÷63°С/10 Торр. ЯМР 1H: δ 2,2 (c, CH3); ЯМР 19F: δ -65 (с, CF3).
1,1,1-Трихлор-3,3,3-трифтор-2-(трифторметил)пропилтрифторацетат (4b).
К смеси 200 г (0,7 моль) карбинола 2 и 220,5 г (1,05 моль) (CF3CO)2O при перемешивании и температуре 5÷10°С медленно по каплям добавляют 77,9 г (0,77 моль) триэтиламина. Смесь перемешивают в течение 2-3 часов при 20÷25°С. Отгоняют летучие компоненты смеси в вакууме (10-15 Торр) в охлаждаемую сухим льдом ловушку, затем фракцию, выкипающую при 50÷60°С/10-15 Торр. Повторной перегонкой дистиллята получают 227 г (85%) эфира 4b, т.кип. 60°С/10 Торр. ЯМР 19F δ: -77 (с, 3F, CF3CO2), -65,8 (с, 6F, (CF3)2С).
1,1,1-Трихлор-3,3,3-трифтор-2-(трифторметил)пропилметансульфонат (4c).
К раствору 427,5 г (1,498 моль) карбинола 2 в 1 л ацетонитрила при перемешивании и температуре 5÷10°С прибавляют 167 г (1,65 моль) триэтиламина и добавляют по каплям 189 г (1,65 моль) метансульфонилхлорида. Смесь перемешивают 3-4 часа при температуре 20÷25°С и оставляют на ночь. Затем смесь выливают в двукратный объем воды, нижний слой отделяют, промывают два раза двукратным объемом воды, при необходимости добавляя немного ацетона для предотвращения загустевания, добавляют 10 г P2O5 и перегоняют в вакууме (0,1 Торр) собирая фракцию, выкипающую при 60÷80°С.
Получают 508,3 г (93%) сульфоната 4c, содержащего 4% 2. Т.кип. 80°С/0,1 Торр. ЯМР 1H: δ 3,35 (c, CH3); ЯМР 19F: δ -64,5 (с, CF3).
1,1-Дихлоргексафторизобутилен (1).
К суспензии 108 г (1,65 моль) Zn-пыли в 300 мл сульфолана, активированной 8 мл триметилхлорсилана, при перемешивании и температуре 150÷160°С добавляют по каплям 300 г (0,825 моль) метансульфоната 4c с одновременной отгонкой образующегося олефина 1, содержащего 2-8% 1-хлоргексафторизобутилена 5 и следовые количества ГМДС. Полученный продукт перегоняют из конц. H2SO4 и при необходимости ректифицируют.
Получают 153,8 г (80%) олефина 1, т.кип. 74°С (лит. 71÷74°С [30]). ЯМР 19F: δ -61,5 (с, CF3).
1-Хлоргексафторизобутилен (5): ЯМР 1H: δ 5,4 (уш.c, H); ЯМР 19F: δ -66,4 (м, 3F, CF3), -63 (м, 3F, CF3). Масс-спектр (M/Z, отнесение): 198[M]+, 179[M-F]+, 163[M-Cl]+, 147[M-HCF2]+, 131[M-Cl-HCF]+, 129[M-CF3]+, 69[CF3]+(100%), 67[HCFCl]+, 48[CHCl]+.
Общая методика синтеза соединений 7a-с.
К суспензии 100 г (0,658 моль) сухого фтористого цезия в 150 мл диметилформамида при перемешивании и температуре 15÷20°С прибавляют 42 г (0,18 моль) дихлоргексафторизобутилена (1). Реакционную смесь перемешивают в течение 2 часов, контролируя температуру смеси внешним охлаждением, чтобы она не повышалась выше 30°С. Далее при перемешивании и температуре 20÷25°С прибавляют 0,2 моль аллилбромида или бромэтилацетата, или 0,1 моль полухлористой серы (S2Cl2). Смесь перемешивают в течение 2-3 часов и оставляют на ночь. Затем реакционную смесь разбавляют 350 мл 5% соляной кислоты, продукт экстрагируют CH2Cl2, полученный раствор промывают несколько раз 5% соляной кислотой, растворитель отгоняют и остаток перегоняют при атмосферном давлении или в вакууме. Дополнительную очистку продукта осуществляют ректификацией.
(CF3)3CCH2CH=CH2 (7a).
Получают 32,8 г (70%), т.кип. 76÷77°С (лит. 75÷76°С [31]). ЯМР 19F: δ -67 (с, CF3).
(CF3)3CCH2CH2OAc (7b).
Получают 38,5 г (70%), т.кип. 141÷142°С (лит. 140÷142°С [32]). ЯМР 19F: δ -69 (с, CF3).
(CF3)3CSSSC(CF3)3 (7c).
Получают 28,8 г (60%), т.кип. 160°С (лит. 96÷100°С/50 Торр [33-34]). ЯМР 19F: δ -65 (с, (CF3)3C).
(CF3)3CBr (7d).
К суспензии 100 г (0,658 моль) сухого фтористого цезия в 150 мл ацетонитрила при перемешивании и температуре 15÷20°С прибавляют 42 г (0,18 моль) олефина 1. Реакционную смесь перемешивают в течение 4-5 часов, контролируя температуру смеси внешним охлаждением, чтобы она не повышалась выше 30°С. Далее при перемешивании и температуре 5÷10°С прибавляют 28,8 г (0,18 моль) брома. Смесь перемешивают в течение 15 минут и нагревают с отгонкой до температуры кипения 70°С, собирая дистиллят в охлаждаемый (0°С) приемник. Полученный раствор оставляют стоять в холодильнике (~-20°С) в течение ночи, образовавшийся кристаллический продукт отделяют, смешивают на холоду с равным объемом конц. H2SO4 и возгоняют.
После повторной возгонки получают 32,3 г (60%) бромида 7d, чистотой 90%, т.сублимации 35÷40°С (лит. т.пл. 58÷59°С [35]). ЯМР 19F: δ -68 (с, (CF3)3C).
(E)-1-Бром-6,6,6-трифтор-5,5-бис(трифторметил)гекс-2-ен (7e).
К суспензии 200 г (1,32 моль) сухого фтористого цезия в 400 мл ДМФА при перемешивании и температуре 15÷20°С прибавляют 84 г (0,36 моль) олефина 1. Реакционную смесь перемешивают в течение 2 часов, контролируя температуру смеси внешним охлаждением, чтобы она не повышалась выше 30°С. Далее при перемешивании и температуре 20÷25°С присыпают 92 г (0,43 моль) 1,4-дибромбут-2-ена. Смесь перемешивают в течение 2-3 часов и оставляют на ночь. Затем реакционную смесь разбавляют 700 мл 5% соляной кислоты, продукт экстрагируют CH2Cl2, полученный раствор промывают несколько раз 5% соляной кислотой, растворитель отгоняют и остаток перегоняют в вакууме (10 Торр) собирая фракцию, выкипающую при 40÷60°С. Продукт повторно перегоняют в вакууме, собирая фракцию с т.кип. 50÷55°С/10 Торр. Полученный дистиллят оставляют в холодильнике (~5°С) в течение ночи, выпавшие кристаллы исходного 1,4-дибромбут-2-ена отфильтровывают и получают 106 г 7e, который используют в последующем синтезе без дополнительной очистки.
(E)-6,6,6-Трифтор-5,5-бис(трифторметил)гекса-1,3-диен (8).
Смесь 100 г (0,28 моль) 7e и 31,9 г (0,57 моль) гидроксида калия в 150 мл сульфолана нагревают при перемешивании до начала сильной экзотермической реакции, в результате которой смесь начинает интенсивно кипеть. После завершения самопроизвольного кипения реакционной смеси, ее нагревают до кипения при перемешивании в течение 30 минут и продукт реакции отгоняют, собирая фракцию кипящую 80÷100°С. После отделения воды, продукт перегоняют над KOH и очищают ректификацией.
Получают 45,7 г (60%) 8, чистотой 98+% по ГЖХ, т.кип. 97°С. Найдено (%): C, 35,38; H, 1,84; F, 63,08. C8H5F9. Вычислено (%): C, 35,31; H, 1,85; F, 62,84. ЯМР 19F: δ -68 (с, CF3).
Реакция олефина 1 с МеОН в присутствии оснований; получение эфиров 9 и 19.
К раствору 2,83 г KOH в 12 мл МеОН постепенно при перемешивании прибавили 10 г (43 ммол) олефина 1, по окончании экзотермической реакции перемешивали 0,5 часа, затем реакционную массу промыли водн. HCl, органический слой отделили, высушили над MgSO4 и получили 8 г смеси, содержащей, по данным хроматомасс-спектроскопии, 84% эфира 9 (спектр ЯМР 19F идентичен описанному в [36]), выход 76% с учетом конверсии олефина 1, 12% олефина 1 и 4% эфира 19. Реакция 1 с MeOH в присутствии KF приводит к образованию аналогичной по составу смеси продуктов.
Масс-спектр 9 (M/Z, отнесение): 228[M]+; 209[M-F]+; 193[M-Cl]+, 178[C4F6O]+; 159[C4F5O]+; 150[C3F6]+; 129[C3HF4O]+; 109[C3F3O]+; 91[C3HFCl]+; 81[C2F3]+; 71[C3FO]+; 69[CF3]+; 63[CClO]+.
Обработка полученной смеси конц. H2SO4 приводит к количественному превращению эфира 9 в метиловый эфир α-гидроперфторизомасляной кислоты 19 (по данным ЯМР 19F-спектроскопии и хромато-масс-спектрометрии).
2,2-Дихлор-3,3-бис(трифторметил)оксиран (10).
К 50 г (0,175 моль) кабинола 2 добавляют 14,56 г (0,26 моль) гидроксида калия, смесь перемешивают в течение 0,5-1 часа и медленно нагревают до начала экзотермической реакции, в процессе которой смесь интенсивно закипает. Кипение поддерживают в течение 30 минут и продукт реакции отгоняют.
Полученный дистиллят, после отделения воды, перегоняют над P2O5 и получают 35 г (87%) оксирана 10, т.кип. 69÷70°С (лит. 68÷70°С [12]). ЯМР 19F: δ -69,4 (с, CF3).
3-Хлор-2,2-бис(трифторметил)оксиран (11).
К 69 г (0,275 моль) карбинола 3 добавляют при перемешивании 23 г (0,41 моль) гидроксида калия, через несколько минут начинается экзотермическая реакция и смесь разогревается. Температуру смеси контролируют внешним охлаждением так, чтобы она не закипала. После окончания экзотермической реакции смесь кипятят в течение 30 минут и продукт реакции отгоняют.
Полученный дистиллят перегоняют над P2O5 и получают 48 г (81%) оксирана 11, т.кип. 52÷53°С. ЯМР 1H: δ 5,4 (уш.c, H); ЯМР 19F: δ -75,2 (м, 3F, CF3), -68,6 (м, 3F, CF3).
2,3,3,3-Тетрафтор-2-(трифторметил)пропаноилфторид (13).
Смесь 16,27 г (0,28 моль) сухого KF и 20 г (0,07 моль) карбинола 2 в 50 мл абс. диглима медленно нагревают при перемешивании с обратным холодильником до начала экзотермической реакции, в процессе которой газообразные продукты реакции отгоняются из реакционной смеси и собираются в охлаждаемую (-78°C) ловушку. Содержимое ловушки перегоняют, собирая легкую фракцию.
Получают 7,5 г (50%) 13, т.кип. 2÷4°С (лит. 2÷3°С [37]). ЯМР 19F δ: -183,4 (м, 1F, CF(CF3)2), -77,2 (д, 6F, 3JFF = 5,6 Гц, CF(CF3)2), 29,6 (м, 1F, COF).
2-Хлор-3,3,3-трифтор-2-(трифторметил)пропаноилхлорид (14).
К суспензии 0,76 г (0,013 моль) сухого фтористого калия в 30 мл сульфолана при перемешивании прибавляют 67 г (0,27 моль) оксирана 10. Смесь нагревают при перемешивании до кипения в течение 30 минут и продукт реакции отгоняют с колонкой Вигре. Полноту изомеризации 10 в 14 контролируют ЯМР 19F: 10 (δ: -67,5); 14 (δ: -70,1).
Получают 60,5 г (90%) 14, т.кип. 68÷69°С (лит. 70°С [5]). ЯМР 19F δ: -70 (с, CF3).
2-Хлор-3,3,3-трифтор-2-(трифторметил)пропаноилфторид (15).
Смесь 60 г (0,241 моль) 14 и 35 г (0,6 моль) сухого KF в 30 мл сульфолана нагревают до кипения при перемешивании с колонкой Генкеля и насадкой полной конденсации, постепенно отбирая дистиллят с т.кип. 35÷50°С. Полноту превращения 14 в 15 контролируют ЯМР 19F: 14 (δ: -70,2); 15 (δ: -71,5).
Получают 42 г (75%) 15, т.кип. 34÷35°С (лит. 34°С [5]). ЯМР 19F δ: -71,7 (д, 6F, 4JFF = 11 Гц, CF3), 32,4 (м, 1F, COF).
Метил-2,3,3,3-тетрафтор-2-(трифторметил)пропаноат (16).
К суспензии 28 г (0,48 моль) сухого KF в 100 мл метанола при перемешивании прибавляют 40 г (0,16 моль) оксирана 10. Смесь разогревается до кипения, после завершения экзотермической реакции, смесь кипятят в течение 1 часа, охлаждают, выливают в холодную воду, нижний слой промывают холодной водой и перегоняют над P2O5. Продукт очищают ректификацией.
Получают 29 г (80%) 16, т.кип. 75÷76°С (лит. 76÷77°С [38]). ЯМР 1H: δ 4,3 (уш.с, OCH3); ЯМР 19F δ: -183 (c, 1F, CF(CF3)2), -76,5 (с, 6F, CF(CF3)2).
Реакция 2,2-дихлор-3,3-бис(трифторметил)оксирана 10 с KBr.
К суспензии 14,3 г (120 ммол) KBr в 50 мл диглима постепенно прибавляют 10 г (40 ммол) оксирана 10, перемешивают до окончания экзотермической реакции, затем перемешивают при 75÷83°С/1,5 часа. По данным ЯМР 19F реакционная масса содержит смесь галогенангидридов α-бром-, α-хлор- и α-гидроперфторизомасляных кислот в мольном соотношении 4:2:1 соответственно. К реакционной массе прибавили 8 мл МеОН, перемешивали 10 мин, промыли разб. HCl-кислотой, органический слой отделили и получили 9,95 г смеси, содержащей 90% эфиров (CF3)2CBrCO2Me (17) : (CF3)2CClCO2Me (18) : (CF3)2CHCO2Me (19) в мольном соотношении 1:1:0,8.
Масс-спектр эфира 17 (M/Z, отнесение): 288[M]+; 257[C4BrF6O]+; 229[C3BrF6]+; 210[C3BrF5]+; 191[C3BrF4]+; 179[C2BrF4]+; 178[C4F6O]+; 169[C3BrF2O]+; 160[C2BrF3]+; 159[C4H3F4O2]+; 150[C3F6]+; 131[C3F5]+(100%); 129[CBrF2]+; 112[C3F4]+; 100[C2F4]+; 93[C3F3]+; 81[C2F3]+; 71[C3FO]+, 69[CF3]+; 59[C2H3O2]+; 47[CFO].
Масс-спектр эфира 18 (M/Z, отнесение): 213[C4ClF6O]+; 185[C3ClF6]+; 166[C3ClF5]+; 159[C4H3F4O2]+; 150[C3F6]+; 131[C3F5]+; 116[C2ClF3]+; 100[C2F4]+; 85[CClF2]+; 81[C2F3]+; 69[CF3]+(100%); 59[C2H3O2]+; 47[CFO].
Масс-спектр эфира 19 (M/Z, отнесение): 225[M+Me]+; 211[M+H]+(100%); 191[M-F]+; 179[C4HF6O]+; 170[C5H2F4O2]+; 159[C4H3F4O2]+; 150[C3F6]+; 140[C4H3F3O2]+; 113[C3HF4]+; 91[C3HF2O]+; 81[C2F3]+; 69[CF3]+; 59[C2H3O2]+.
Реакция оксирана 10 с 0,5 экв. SbF5.
2-Хлор-2-фтор-3,3-бис(трифторметил)оксиран (21).
К 30 г (0,12 моль) оксирана 10 добавляют порциями при перемешивании и температуре 20÷25°С 13 г (0,06 моль) SbF5. Смесь перемешивают в течение часа, выливают на колотый лед, добавляют соляной кислоты, нижний слой отделяют, промывают разбавленной соляной кислотой и перегоняют из H2SO4(конц.).
Получают 19 г дистиллята, содержащего по данным ГЖХ и ЯМР 19F-спектроскопии 25% окиси 21 и 63% карбинола 12.
В следующем опыте аналогично полученную реакционную смесь медленно нагревают с одновременной отгонкой продукта реакции в охлаждаемый сухим льдом приемник. Получают 25,5 г дистиллята, содержащего по данным ГЖХ и ЯМР 19F 89% оксирана 21, который очищают ректификацией.
Получают 22 г (80%) оксирана 21, т.кип. 34,5÷35,5°С. Найдено (%): С, 20,53; Cl, 14,68; F, 56,73. C4СlF7O. Вычислено (%): С, 20,67; Cl, 15,25; F, 57,20. ЯМР 19F δ: -88 (м, 1F, CFCl), -70 (c, 6F, CF3). Масс-спектр (M/Z, отнесение): 216[M-O]+, 197[M-Cl]+, 181[C4F7]+, 169[C3F7]+, 147[C3ClF4]+, 119[C2F5]+, 93[C3F3]+, 85[CClF2]+, 69[CF3]+(100%), 47[CFO]+.
Масс-спектр карбинола 12 (M/Z, отнесение): 252[M]+, 233[M-F]+, 217[M-Cl]+, 197[C4F7O]+, 185[C3F6Cl]+, 163[C3ClF4O]+, 147[C3F5O]+, 119[C2F5]+, 85[CClF2]+, 69[CF3]+(100%), 51[CHF2]+, 50[CF2]+.
Реакция окиси 10 с 1 экв. SbF5.
Вариант 1. К 30 г (0,12 моль) оксирана 10 добавляют порциями при перемешивании и температуре 20÷25°С 26 г (0,12 моль) SbF5. Далее вязкую, медообразную смесь перемешивают в течение часа, выливают на лед, добавляют соляной кислоты, нижний слой отделяют, промывают разбавленной соляной кислотой и перегоняют из H2SO4(конц.).
Получают 26 г (85%) спирта 12, т.кип. 74÷75°С (лит. 73÷74°С [29]). ЯМР 1H: δ 3,7 (уш.с, OH); ЯМР 19F δ: -74,3 (т, 6F, 4JFF = 7,5 Гц, CF3), -62,2 (септ., 2F, CF2Cl).
Вариант 2. Аналогично полученную реакционную смесь медленно нагревают до температуры 180÷190°С с одновременной отгонкой продуктов реакции в охлаждаемый сухим льдом приемник. Получают 19 г дистиллята, содержащего по данным ЯМР 19F: 36% окиси 21; 33% окиси перфторизобутилена (22) и 29% фторангидрида 13, а также незначительные примеси других продуктов реакции, как например 2% спирта 12, и хлора.
Литература
- Yu.V. Zeifman, E.G. Ter-Gabrielyan, N.P. Gambaryn, I.L. Knunyants Russ.Chem.Rev., 1984, 53, 256-273.
- N.P. Gambaryan, E.M. Rokhlin Russ.Chem.Rev., 1986, 55, 480-494.
- I.L. Knunyants, V.R. Polishchuk Russ.Chem.Rev., 1976, 45, 574-592.
- B.L. Dyatkin, N.I. Delyagina, S.R. Sterlin Russ.Chem.Rev., 1976, 45, 607-614.
- I.L. Knunyants, Yu.A. Cheburkov, M.D. Bargamova Bull.Acad.Sci.USSR, Div.Chem.Sci., 1963, 12, 1265-1268.
- D.C. England, C.G. Krespan J.Am.Chem.Soc., 1965, 87, 4019-4020.
- M.S. Raasch J.Org.Chem., 1970, 35, 3470-3483.
- E.M. Rokhlin, E.G. Abduganiev, U. Utebaev Russ.Chem.Rev., 1976, 45, 593-606.
- S. Ebnesajjad Fluoroplastics, Vol. 1, 2nd Ed., pp. 48-75, 2015.
- C.M. Timperley Fluorine Chemistry at the Millennium Fascinated by Fluorine, Ed. R.E. Banks, pp. 499-538, 2000.
- C.M. Timperley J.Fluor.Chem., 1999, 94, 37-41.
- Yu.V. Zeifman Bull.Russ.Acad.Sci., Div.Chem.Sci., 1992, 41, 370-373.
- A.A. Tyutyunov, A.V. Sin’ko, S.M. Igumnov, O.A. Mel’nik, Ya.S. Vygodskii, E.V. Khaidukov, V.I. Sokolov Dokl.Chem., 2016, 467, 88-90.
- R.E.A. Dear, E.E. Gilbert, J.J. Murray Tetrahedron, 1971, 27, 3345-3355.
- H.B. Dykstra, J.F. Lewis, C.E. Boord J.Am.Chem.Soc., 1930, 52, 3396-3404.
- J.W. Cornforth, R.H. Cornforth, K.K. Mathew J.Chem.Soc., 1959, 112-127.
- R.A. Bekker, G.V. Asratyan, B.L. Dyatkin, I.L. Knunyants Dokl.Akad.Nauk SSSR, 1972, 204, 606-609.
- R.A. Bekker, G.V. Asratyan, B.L. Dyatkin Zhur.Org.Khim., 1973, 9, 1635-1640.
- A.N. Nesmeyanov, K.A. Pecherskaya, G.Ya. Uretskaya Izv.Acad.Nauk USSR, Otdel.Khim.Nauk, 1948, 240-245.
- J.H. Clark Chem.Rev., 1980, 80, 429-454.
- W. Reeve Synthesis, 1971, 131-138.
- T.S. Snowden ARKIVOC, 2012, 2, 24-40.
- Yu.A. Cheburkov, M.D. Bargamova Bull.Acad.Sci.USSR, Div.Chem.Sci., 1967, 16, 801-806.
- I.L. Knunyants, V.V. Shokina, V.V. Tyuleneva, T.N. Razumeeva Bull.Acad.Sci.USSR, Div.Chem.Sci., 1972, 21, 1085-1088.
- J.T. Hill J.Fluor.Chem., 1977, 9, 97-112.
- D.E. Morin US Pat. № 3,213,134 (1965).
- F.J. Pavlik US Pat. № 3,385,904 (1968).
- R. Filler, R.M. Schure J.Org.Chem., 1967, 32, 1217-1219.
- R.E.A. Dear Synthesis, 1970, 361-362.
- V.V. Tyuleneva, L.A. Rozov, Yu.V. Zeifman, I.L. Knunyants Bull.Acad.Sci.USSR, Div.Chem.Sci., 1975, 24, 1042-1045.
- N.I. Delyagina, E.Ya. Pervova, I.L. Knunyants Bull.Acad.Sci.USSR, Div.Chem.Sci., 1972, 21, 326-329.
- I.L. Knunyants, N.I. Delyagina, B.L. Dyatkin, I.Ya. Aliev USSR certificate of authorship № 379556 (1973).
- C.G. Krespan, D.C. England J.Org.Chem., 1968, 33, 1850-1854.
- D.C. England US Pat. № 3,544,591 (1970).
- B.L. Dyatkin, A.A. Gevorkyan, I.L. Knunyants Bull.Acad.Sci.USSR, Div.Chem.Sci., 1965, 14, 1833-1835.
- D.I. Rossman, A.J. Muller J.Fluor.Chem., 1993, 60, 61-68.
- I.L. Knunyants, V.V. Shokina, V.V. Tyuleneva, Yu.A. Cheburkov, Yu.E. Aronov Bull.Acad.Sci.USSR, Div.Chem.Sci., 1966, 15, 1764-1766.
- R.D. Smith, F.S. Fawcett, D.D. Coffman J.Am.Chem.Soc., 1962, 84, 4285-4288.
Статья рекомендована к публикации членом редколлегии к.х.н. В.В. Корниловым
Fluorine Notes, 2018, 121, 9-10


