Поступило в редакцию: Ноябрь 2017
УДК 542.91:541.49+541.64:66.095.264.3-036.742
Fluorine Notes, 2018, 117, 7-8
Бис-салицилидениминные фторсодержащие комплексы титана(+4) в полимеризации высших -олефинов
С. Ч. Гагиева1*, В.А. Тускаев1, О.В.Смирнова2, К.Ф. Магомедов3, Б. М. Булычев1
*1Московский государственный университет имени М.В. Ломоносова. Химический факультет, 119992, Москва, Ленинские горы
2 Казанский государственный университет
3Российский химико-технологический университет имени Д.И. Менделеева (D.Mendeleev University of Chemical Technology of Russia)
Автор для контактов: С.Ч. Гагиева (E-mail: sgagieva@yandex.ru)
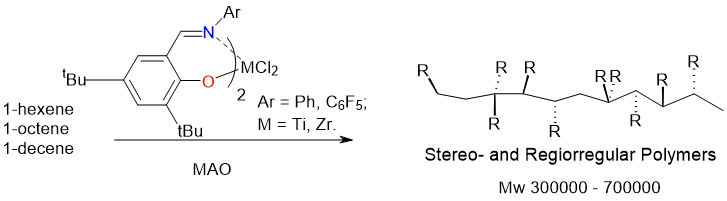
Аннотация: Изучены каталитические свойства постметаллоценовой системы на основе комплекса титана(+4) и циркония(+4) с бис-салицилидениминным фторсодержащим лигандом в полимеризации высших олефинов. Показано, что активация комплекса метилалюмоксаном (МАО) в зависимости от условий синтеза приводит к образованию синдио- и изотактических поли--олефинов со значениями молекулярных масс от 0.01*105 до 19.98*105 и с продуктивностью до 2217 кг Полимера/моль Ti).
ВВЕДЕНИЕ
В конце 1990-х годов появились сообщения [1,2] о синтезе новых высокоактивных катализаторов полимеризации олефинов на основе комплексов металлов четвёртой группы с бис(феноксииминовыми) и бис(пирролилиминовыми) лигандами. В зависимости от структуры пре-катализаторов активность постметаллоценовых систем достигала и даже превышала активность металлоценовых систем. Некоторые комплексы оказались способны катализировать процессы «живой» полимеризации этилена, стереоспецифичной полимеризации пропилена и даже сополимеризации этилена и α-олефинов с полярными сомономерами [3].
Одно из самых интересных свойств катализаторов серии FI связано с возможностью варьировать электронные и стерические характеристики пре-катализатора путем подбора лигандов с различными заместителями (“настройка” состава и структуры катализатора на синтез полиолефина с определенными свойствами) Кроме того, было показано, что некоторые FI комплексы титана, как правило, с фторсодержащими заместителями у иминного фрагмента лиганда при их активации полиметилалюмоксаном (МАО) способны проводить “живую” полимеризацию этилена и пропилена, в том числе и при повышенных температурах (50-70 °С) [1,4-14]. Термин “живая” полимеризация подразумевает линейную зависимость молекулярной массы полимера от времени процесса, то есть наличие в системе каталитических центров одного типа. Механизм данного эффекта не может быть объяснён исключительно электроакцепторным эффектом атомов фтора, повышающим электрофильность металлического центра и приводящим к снижению энергии активации внедрения молекулы этилена, как предлагалось в работах [13-14]. В настоящее время экспериментально доказано методами ЯМР-спектроскопии и нейтронной дифрактометрии, а также подтверждено DFT-расчетами, что атом фтора лиганда взаимодействует с атомом водорода в β-положении полимерной цепи, предотвращая процесс β-гидридного переноса [15]. Комплексы с атомами фтора только в мета- и/или пара-положениях анилинового кольца также высокоактивны, однако не являются катализаторами «живой» полимеризации (Mw/Mn=1.78-2.18) [16-17]. Возможность осуществления «живой» полимеризации этилена и пропилена продемонстрирована в [18] на примере биядерных фторсодержащих бис(салицилиден)иминных комплексов титана.
Однако использование феноксииминных катализаторов в гомополимеризации высших олефинов с МАО в качестве сокатализатора изучено в гораздо меньшей степени (см. [19]. В данной работе приводятся результаты изучения полимеризации высших -олефинов (гексена-1, октена-1 и децена-1) на постметаллоценовых комплексах, представленных на схеме 1.
Схема 1
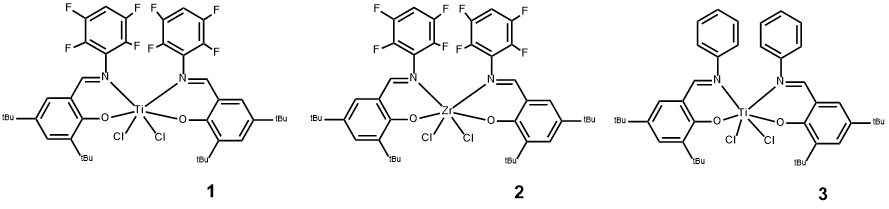
Изучение каталитической активности каталитических систем с участием комплексов (прекатализаторов) 1-3 проводили в массе -олефинов при 25°C с использованием в качестве активатора коммерческого полиметилалюмоксана (МАО). Мольное отношение сокатализатор/прекатализатор варьировали в диапазоне 100-1000 моль/моль при постоянной концентрации прекатализаторов 2.3*10-6 моль/л. Относительно низкая активность системы при 25°C определила необходимость проводить полимеризацию в течение длительного времени (10 суток). Основные результаты, полученные при этом исследовании, суммированы в Таблице 1.
Таблица 1. Каталитическая активность 1-3 в полимеризации -олефинов (С6 – гексен-1, С8 – октен-1, С10 – децен-1) в массе (объем мономера – 9 мл, [Ti] = 2.3·10-6 моль/л, сокатализатор – МАО, температура 25°С), время полимеризации 10 дней.
|
No |
Прекатализатор |
Мономер |
Al/Ti |
Выход, г |
A, кг ПОЛ/моль Ti |
Mw , 105 |
Mn, 104 |
Mw/Mn |
|
1 |
1 |
С6 |
100 |
0.4 |
182.6 |
8.72 |
4.13 |
21.1 |
|
2 |
1 |
С6 |
250 |
5.1 |
2217.4 |
5.90 |
5.71 |
10.3 |
|
3 |
1 |
С6 |
500 |
5.0 |
2173.9 |
6.19 |
4.58 |
13.3 |
|
4 |
1 |
С6 |
750 |
4.5 |
1956.5 |
8.17 |
3.71 |
22.1 |
|
5 |
1 |
С6 |
1000 |
4.6 |
2000.0 |
19.98 |
1.41 |
141.9 |
|
6 |
2 |
С6 |
1200 |
2.2 |
349.2 |
0.09 |
0.12 |
7.9 |
|
7 |
2 |
С6 |
1500 |
2.2 |
349.2 |
0.01 |
0.09 |
1.4 |
|
8 |
3 |
С6 |
250 |
2.5 |
929.6 |
1.19 |
2.21 |
5.4 |
|
9 |
3 |
С6 |
500 |
0.9 |
314.8 |
0.99 |
2.16 |
4.6 |
|
10 |
3 |
С6 |
750 |
0.3 |
118.5 |
1.28 |
2.06 |
6.2 |
|
11 |
3 |
С6 |
1000 |
0.1 |
37.0 |
3.31 |
1.62 |
20.4 |
|
12 |
1 |
С8 |
100 |
0.6 |
265.2 |
5.81 |
4.28 |
13.6 |
|
13 |
1 |
С8 |
250 |
1.5 |
652.2 |
5.72 |
6.47 |
8.8 |
|
14 |
1 |
С8 |
500 |
2.0 |
856.5 |
5.83 |
6.52 |
8.9 |
|
15 |
1 |
С8 |
750 |
1.8 |
795.7 |
5.49 |
6.24 |
8.8 |
|
16 |
1 |
С8 |
1000 |
1.8 |
782.6 |
5.10 |
5.09 |
9.9 |
|
17 |
2 |
С8 |
560 |
1.9 |
293.6 |
0.02 |
0.11 |
1.4 |
|
18 |
2 |
С8 |
1200 |
6.5 |
663.3 |
0.02 |
0.13 |
1.5 |
|
19 |
2 |
С8 |
1500 |
6.5 |
1000.0 |
0.02 |
1.29 |
1.5 |
|
20 |
2 |
С8 |
2000 |
4.0 |
634.9 |
0.02 |
0.12 |
1.6 |
|
21 |
1 |
С10 |
100 |
0.6 |
239.1 |
4.11 |
2.61 |
15.8 |
|
22 |
1 |
С10 |
250 |
1.2 |
521.7 |
5.31 |
6.53 |
8.1 |
|
23 |
1 |
С10 |
500 |
1.7 |
734.8 |
4.63 |
4.92 |
9.7 |
|
24 |
1 |
С10 |
750 |
1.2 |
500.0 |
5.31 |
5.81 |
9.1 |
|
25 |
1 |
С10 |
1000 |
1.4 |
595.6 |
5.09 |
3.89 |
13.0 |
|
26 |
3 |
С10 |
250 |
0.6 |
237.0 |
0.01 |
0.01 |
7.9 |
|
27 |
3 |
С10 |
500 |
2.0 |
722.2 |
0.06 |
0.09 |
6.5 |
|
28 |
3 |
С10 |
750 |
2.1 |
777.8 |
0.04 |
0.08 |
4.9 |
|
29 |
3 |
С10 |
1000 |
2.2 |
814.8 |
0.09 |
0.09 |
9.3 |
|
30 |
3* |
С6 |
500 |
0.3 |
120.0 |
0.55 |
1.91 |
2.9 |
* к катализатору добавили МАО, выдерживали неделю, после добавили гексен-1
Как видно из таблицы 1, c увеличением мольного соотношения Al/Ti от 250 до 1000 при использовании комплекса 1 происходит незначительное падение активности от 2217.4 до 2000 кг полимера/моль Ti и одновременно существенное увеличение как средневесовой молекулярной массы полигексена почти до 2*106, так и значения молекулярно-массового распределения до 121. В то же время в случае применения в системе аналогичного по составу комплекса циркония(+4) (2) увеличение мольного соотношения Al/Ti приводит к резкому снижению активности, молекулярной массы и ММР. Причем последнее понижается до значения 1.4, т.е. система становится одноцентровой (с одним типом активного центра).
Каталитическая система, сформированная на прекатализаторе 3, в лиганде которого нет атомов фтора, также показывает своеобразные свойства. В этом случае увеличение отношения Al/Ti от 250 до 1000 сразу приводит к падению активности до минимального в ряду этих систем значения – 37 кг Полигексена/моль Ti, хотя разброс значений ММР в этом случае заметно меньше, чем наблюдаемый на системе 1.
Очень высокие значения ММР полимеров, полученных на системах с прекатализаторами 1 и 3, указывают на их многоцентровость. Этому могут быть несколько причин, начиная от различий в природе металла и свойств его соединений (например, легко восстанавливаемый при действии алюмиийорганических соединений атом титана(+4) и устойчивый к этому воздействию атом циркония(+4)) и кончая свойствами активатора - коммерческого МАО, содержащего от 30 до 40 моль % сопутствующего триметилалюминия (ТМА), и способом формирования системы. Действительно, если после смешения прекатализатора 1 с коммерческим МАО выдержать смесь в течение нескольких дней и затем ввести в раствор гексен-1, то обнаруживается, что активность системы уменьшается почти в 18 раз (ср. опыт 3 в «стандартном» исполнении с результатами опыта 30, таблица 1) и молекулярная масса также уменьшается на порядок.
Исходя из этих данных, можно предположить, что в результате длительной обработки алюминийорганическими соединениями (ТМА и МАО) атом титана(+4) в комплексе 1 восстанавливается до титана(+3) и именно он с ним формируется каталитическая система, которая имеет иное строения и иную активность, чем система с соединениями титана(+4). Это предположение косвенно подтверждают экспериментальные данные по полимеризации этилена на коммерческом МАО и т.н. «сухом» МАО (т.е. МАО из которого длительным вакуумированием практически нацело отогнан триметилалюминий). Нами было показано, что присутствие ТМА в сокатализаторе приводит к снижению активности системы и значительному уменьшению молекулярной массы полимера по сравнению с каталитической системой, активированной “сухим” МАО, что объясняется более эффективной передачей растущей полимерной цепи на алюминийорганическое соединение в первом случае [20].
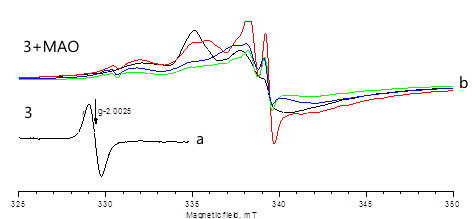
Об образовании титана (+3) свидетельствуют данные ЭПР спектров растворов комплексов, выдержанных с коммерческим МАО несколько суток (рисунок 2, стандарт ТЕМПО). Через два часа количество парамагнитных соединений, содержащих атомы Ti(+3) (g=1.968) составляло 2 моль %, через сутки 50%, через двое – 100% (спектры «b»).
|
Система |
[Ti (+4)], ммоль/л |
[Ti (+3)], ммоль/л через1-2 часа/через сутки |
моль % [Ti (+3)], через1-2 часа/через сутки |
|
3 |
1.1 |
Нет |
- |
|
3+МАО |
3.1 |
¼ |
~50/~100 |
Рис. 3. ЭПР исследования катализатора 3 (кривая а) и катализатора 3 в присутствии МАО (кривая b).
По данным 13С ЯМР спектроскопии микроструктура полученного полигексена соответствует изо- и синдиотактическому полимеру [21]. Содержание изотактических пентад mmmm, определенное по отношению интегральной интенсивности пика 35.21 м.д. к интегральной интенсивности всей пентадной области (33.5-35.4 м.д.), составляет около 12-23 %.
Таким образом, изучены каталитические свойства комплексов титана(+4) с бис-салицилидениминным фторсодержащим лигандом в полимеризации высших олефинов. Показано, что после активации комплексов метилалюмоксаном (МАО) в зависимости от условий синтеза образуются синдио- и изотактических поли--олефины со значениями молекулярных масс от 0.01 105 до 19.98 105 и с продуктивностью до 2217 кг Полимера/моль Ti.
Сделано предположение, что в результате длительной обработки алюминийорганическими соединениями (ТМА и МАО) атом титана(+4) в комплексе 1 восстанавливается до титана(+3), при этом формируется каталитическая система, которая и ведет процесс полимеризации.
Экспериментальная часть
Синтезы комплексов проводили в атмосфере аргона. Тетрагидрофуран, дихлорметан, толуол, изопропанол, гексан и этилацетат марок “х.ч.” дополнительно очищали согласно литературной методике [22]. TiCl4 фирмы Fluka дополнительно перегоняли в атмосфере аргона. Комплексы 1-3 получали в согласии с литературной методикой [23].
Спектры ЯМР растворов лигандов в CDCl3 записывали на приборах “Bruker WP-500” и “Bruker АМХ-400”, ИК спектры - на спектрофотометре “Magna-IR 750”. Элементный анализ проводился на приборах “Carlo Erba-1106” и “Carlo Erba-1108”.
Реакции полимеризации
Полимеризацию высших олефинов осуществляли в главбоксе марки M-Braun в атмосфере сухого аргона, полимеризовали в массе в стеклянной колбе без перемешивания. Активность катализаторов при полимеризации определяли по выходу полимера, отнесенного к молю Ti.
Анализ полимеров
Микроструктуру полимеров исследовали методом ЯМР 13С спектроскопии. Спектры *ЯМР 13С 10%-ных растворов полимеров в смеси (1,2,4-трихлорбензол +дейтерированный бензол) записывали на приборе “Bruker AVANCE III 400” (частота 100.613 МГц) при 120ºC. Отнесение сигналов было основано на литературных данных [24, 25]. Из спектров ЯМР 13С полимерных образцов определяли содержание стерических пентад. Гель-хроматограммы образцов полимеров получали на хроматографе «Agilent 1200» (детектор испарительный по светорассеянию) с использованием Styragel HMW6E колонки в толуоле при 25ºС. Средние ММ рассчитывали по универсальной калибровочной кривой с использованием ПС стандартов.
*ЯМР 13С спектры представлены в PDF версии статьи.
Работа выполнена при финансовой поддержке РФФИ (грант № 17-03-00234).
Литература
- Small, B. L.; Brookhart, M. J. Am. Chem. Soc. 1998, 120, 7143.
- Britovsek, G. J. P.; Gibson, V. C.; Kimberley, B. S.; Maddox, P. J.; McTavish, S. J.; Solan, G. A.; White, A. J. P.; Williams, D. J. Chem. Commun. 1998, 849.
- S. Matsui, Y. Tohi, M. Mitani, J. Saito, H. Makio, H. Tanaka, T. Fujita. Chem. Lett. 1999, 1065.
- Bianchini, C.; Giambastiani, G.; Rios, G. I.; Mantovani, G.; Meli, A.; Segarra, A. M. Coord. Chem. Rev. 2006, 250, 1391.
- Stoufer, R. C.; Busch, D. H. J. Am. Chem. Soc. 1956, 78, 6016.
- Lions, F.; Martin, K. V. J. Am. Chem. Soc. 1957, 79, 2733.
- Johnson, L. K.; Killian, C. M.; Arthur, S. D.; Feldman, J.; PCT Int. Appl. WO9623010, 1996; Chem. Abstr. 1996, 125, 222773.
- P.G. Cozzi, C. Floriani, A Chiesi-Villa, C. Rizzoli. Inorg. Chem. 1995, 34, 29.
- P.G. Cozzi, E. Gallo, C. Floriani, A Chiesi-Villa, C. Rizzoli. Organometallics, 1995, 14, 4994.
- Laine, T. V.; Klinga, M.; Maaninen, A.; Aitola, E.; Leskela, M. Acta Chem. Scand. 1999, 53, 968-973.
- L.K.Floriany, C.M. Killian, J. Am. Chem:. Soc., 1995, 117, 6414.
- S. Matsui, Y. Tohi, M. Mitani, J. Saito, H. Makio, H. Tanaka, T. Fujita. Chem. Lett. 1999, 1065.
- S. Matsui, M. Mitani, J. Saito, Y. Tohi, H. Makio, H. Tanaka, T. Fujita. Chem. Lett. 1999, 1163.
- J. Saito, M. Onda, S. Matsui, M. Mitani, R. Furuyama, H. Tanaka, T, T. Fujita, Macromol. Rapid Commun., 2002, 23, 1118.
- Chan M. C. W., Weak Attractive Ligand–Polymer and Related Interactions in Catalysis and Reactivity: Impact, Applications, and Modeling, Chemistry – An Asian Journal, 2008, 3, 18–27, DOI: 10.1002/asia.200700226.; Bryliakov K. P., Talsi E. P. Coord. Chem. Rev., 2012, 256, 2994– 3007.
- H. Makio, T. Fujita. Macromol. Symp. 2004, 213, 221.
- United States Patent, 5 698 487, Sacchetti , et al. 1997.
- S.C. Gagieva, T.A. Sukhova, D.B. Savinov, N.M. Bravaya, Y.N. Belokon, B.M. Bulychev. Russian Chemical Bulletin, 2004, 53, 12, 2763-2767.
- Makio H., Terao H., Iwashita A., and Fujita T.. Chem. Rev., 2011, 111, 2363–2449, dx.doi.org/10.1021/cr100294r
- N.M. Bravaya, E.E. Faingol'D, A.N. Panin, E.O. Perepelitsina, S.Ch. Gagieva, V.A. Tuskaev, B.M. Bulychev. Polymer Science - Series B, 2010, 52, 11, 629-636.
- S.Ch. Gagieva, V.A. Tuskaev, B.M. Bulychev, O.V. Smirnova, S.S. Galibeev, N.M. Bravaya. Polymer Science - Series B, 2011, 53, 5-6, 299.
- Органикум. Т.2, М.: Мир, 1992, 472 с.
- S.Ch. Gagieva, T.A. Sukhova, D.V. Savinov, V.A. Optov, N.M. Bravaya, Yu N. Belokon´, B.M. Bulychev. Russian Chemical Bulletin, 2003, 52, 8, 1693-1697.
- T. Asakura, M. Demura, Y. Nishiyama. Macromolecules, 1991, 24, 9, 2334.
- X. Zhao, G. Odian, A.Rossi. J. Polym. Sci., Polym. Chem., 2000, 38, 3802.
Статья рекомендована к публикации членом редколлегии д.х.н. С.М. Игумновым
Fluorine Notes, 2018, 117, 7-8


