Поступило в редакцию: Октябрь 2017
УДК 10.17677/fn20714807.2017.06.02
Fluorine Notes, 2017, 115, 3-4
Фотокаталитичекое радикальное присоединение 1,1,2,2-тетрафтор-1-иодоэтана к алкенам
Svatava Voltrová and Petr Beier*
Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 542/2, 166 10 Prague 6, Czech Republic
E-mail: beier@uochb.cas.cz
Анннотация: Сообщается о фотокаталитическом радикальном присоединении и реакции присоединения-элиминирования терминальных алкенов с 1,1,2,2,-тетрафтор-1-иодоэтана при облучении голубым LED цветом. Фотороредокс катализ с Rhodamine B или Eosin Y обеспечен соответствующими 1,1,2,2,-тетрафтор-1-иодоалканами или 1,1,2,2,-тетрафторалканами с хорошими выходами. Полученнные иодопроизводные были успешно гидродеииодированы с образованием насыщенных 1,1,2,2- тетрафторалканов
Ключевые слова: фотокатилиз, радикальное присоединение, тетрафториодэтан,, LED, Rhodamine B; Eosin Y
Введение
Радикальное перфторалкилирование является популярным методом введения перфторалкильных групп в органические молекулы с целью изменения свойств соединений [1-2]. Традиционно перфторалкил галиды были использованы для генерации перфторалкилиных радикалов, однако, более недавно метал сульфонаты и сульфонилхлориды дали подтверждение, что они могут быть подходящими радикальными прекурсорами [3-7]. Различные подходы для гомолитического расщепления связи углерод-галоген в перфторалкил галидах были рассмотрены, они включают радикальные инициаторы (AIBN [8], Et3B [9], или пероксиды [10]) металлы ( магний [11], железо [1], или медь [12], Na2S2O4 [13], каталитический трифенилфосфин [14], или УФ облучение [15-16].
Недавно, окислительно-восстановительная реакция под действием света и в частности промотированное перфторалкилирование видимым светом [7, 17-20] показало альтернативу перфторалкилированию, промотированному радикальными инициаторами В этом случае органические красители получают преимущество над дорогим рутениевым или иридиевым катализом. Здесь мы сообщаем о каталитическом радикальном присоединении или присоединении-отщеплении соответственно Rhodamine B или Eosin Y с помощью HCF2CF2I к терминальным алкенам. Соединения с тетрафторэтильной группой являются редкими [21-26] и эта работа должна улучшить доступность к ним с целью изучения их свойств.
Результаты и обсуждение
В настоящей работе мы использовали голубой LED свет, испускаемый Rhodamine B [27] или Eosine Y [28] как фотокатализаторов в реакциях радикального присоединения или присоединения-отщепления HCF2CF2I с алкенами. Терминальнй алкен 1 образуя различные функциональные группы (алкил, арил, гидроксил, карбоновую кислоту) претерпевал радикальное присоединение только в эквимолярном соотношении с 1,1,2,2-тетрафтор-1-иодэтаном (2) в присутствии (i-Pr)2NEt и каталитическим Rhodamine B в воде, образуя соответствующий тетрафториодалкан 3 с высоким выходом (Схема 1). Реакция протекала мягко при комнатной температуре, и полная конверсия 1 была достигнута в течении 16 часов (ГХЖ контроль). Интересно, что эксперимент с 3b показал, что зеленый LED свет (525 нм) может быть использован с подобной эффективностью, что и синий цвет. В условиях, показанной на Схеме 1 , гекс-1-ин и цис-стильбен не приводили к продуктам присоединения.
Схема 1
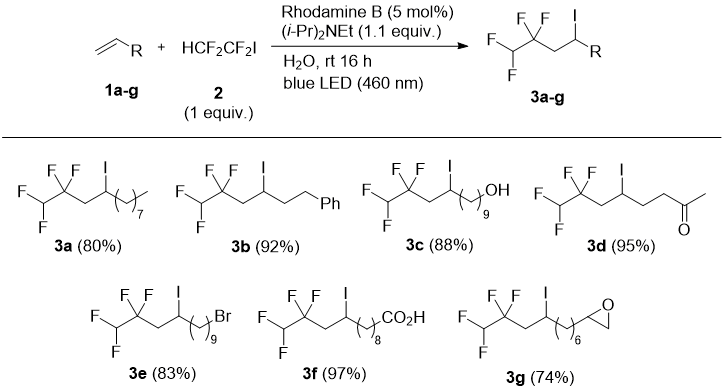
ЯМР 19F спектр иодо производного 3 показал интересное расщепление очевидно по причине присутствия мангитно не эквивалентного (диастереотопного) атома фтора (Рис. 1)
Рис.1. 19F {1H} ЯМР спектр 3a
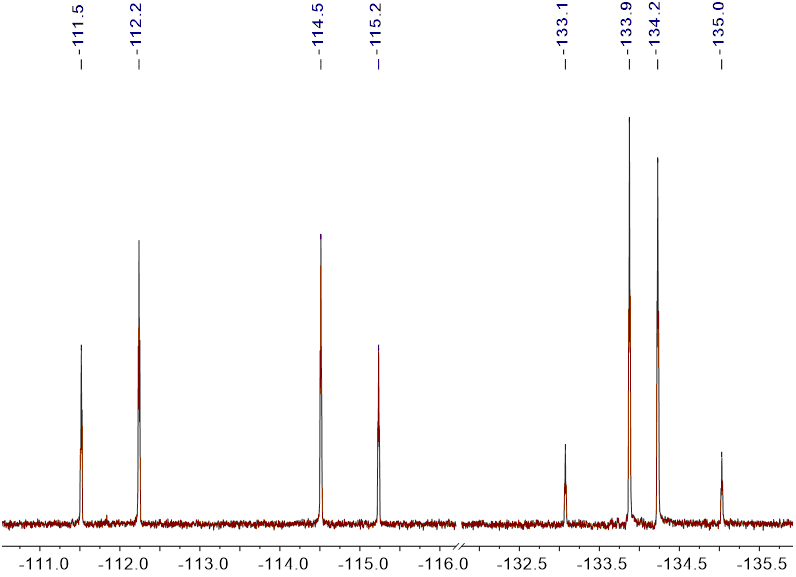
Можно было бы ожидать, что механизм включает гомолитическое расщепление связи углерод-иод с образованием радикала, который добавляется к терминальному алкену. С целью проверки этого механизма реакция с 2,2-диаллилмалонатом (4) была проведена. Образованный фторалкильный радикал вызывал каскад радикальных присоединений – 5-экзо триг циклизацию в циклопентановый продукт 5, который был выделен с хорошим выходом и высокой цис-селективностью (Схема 2)
Схема 2
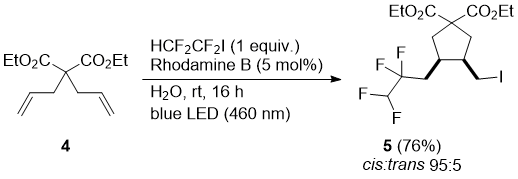
Диэтилаллилмалонат (6) превращался под действием Rhodamine B катализатора главным образом в алкен 7а (продукт элиминирования HI после радикального присоединения) в смеси с продуктом присоединения 7в. Однако применение Eosin Y как фотокатализатора приводило к образованию 7а, что и наблюдатся на схеме 3. Образование продукта элиминирования согласуется с предварительными сообщениями по использованию Eosin Y и карбоната цезия. [28].
Схема 3
В последующем мы приложили наше внимание на замещение атома иода в соединении 3 на водород с целью достигнуть терминальных тетрафторэтил-замещенных алифатичесих соединеий. В направлении этой цели , соединение 3b было обработано избытком LiAlH4 в THF с образованием (5,5,6,6-тетрафторгексил)бензола (8) с высоким выходом ( Схема 4)
Было установлено, что эта реакция зависит от субстрата. Когда эпоксид 3g восстанавливали либо LiAlH или H2/Pd, реакции не наблюдалось даже в течении продолжительного времени (48 ч), и исходный материал выделяли неизменным. 19F ЯМР спектр 8 показывает два набора сигналов: дублет (2JFH = 54.0 Hz) для CF2H группы и триплет дублетов (3JFH = 18.2, 2.0 Hz) для CF2 групппы (Рис. 2).
Схема 4
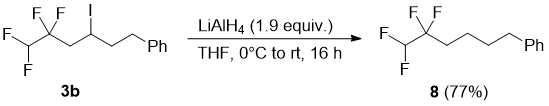
Рис. 2. 19F ЯМР спектр 8; 19F {1H} спектр показан как вставка.
Заключение
Комбинация необычных органических красительных катализаторов и облучение LED приводит к радикальному присоединению тетрафториодэтана к терминальным алкенам, давая 1,1,2,2-тетрафтор-4-иодоалканы с высоким выходом. Альтернативно , радикалное присоединение и замещение и удаление HI обеспечивает соответствующие 1,1,2,2-тетрафторалкены. Производные иода были гидродеиодированы с помощью лития алюминиум гидрида , образуя насыщенные 1,1,2,2-тетрафторалканы. Таким образом, тетрафториодметан (HCF2CF2I) является подезным и ценным реагентом для радикального введения HCF2CF2 группы в фотокаталитических услових.
Экспериментальная часть
Общая процедура.
Реакции с материалами, чувствительными к воздуху, проводили в атмосфере аргона используя стандартную методику Шленка. Были использованы коммерчески доступные реагенты, если не указано иное. Колоночная флэш-хроматография применялась с использованием силикагеля 60 (0.040–0.063 mm). 1H, 13C, и 19F ЯМР спектры были измерены на 5 мм ЯМР трубках. В 13C ЯМР спектрах отвязывались от привязки протонов. Значение химических сдвигов (δ) даны в ppm (м.д.) относительно внутреннего стандарта Me4Si (0 ppm для 1H и 13C ЯМР) или остаточного растворителя и для внутреннего стандарта CFCl3 (0 ppm для 19F ЯМР). Константы взаимодействия (Coupling constants (J)) приведены в Герцах. ГЖХ и масс спектры были получены на 5% метилполисилоксановой колонке, соединенной с квадрупольным масс селективным детектором электронов (70 eV). Масс спектры высокого разрешения (HRMS) были получены с использованием электрораспылительной ионизацией (ESI), электронной ионизацией (EI) или двойной ESI/MALDI ионизацией. Применяли 3 W LED источник (синий 455-475 nm или зеленый 515-535 nm) ,состоящий из светопроводящего стеклянного цилиндра, который был погружен прямо в реакционный сосуд.
Синтез соединения 3. Смесь алкена 1 (1.0 mmol) и воды (10 mL)была охлаждена до 0 °C и дегазирована заполнением с помощью насоса заполнением азотом в течении 3 раз. К этой эмульсии добавляли (i-Pr)2NEt (192 μL, 1.10 mmol), Rhodamine B (24 mg, 0.05 mmol), и HCF2CF2I (227 mg, 103 μL, 1.0 mmol) используя предварительно охлажденный до 0 °C шприц. Перемешиваемая реакционная смесь была нагрета до комнатной температуры и облучалась синей LED лампой в течении 16 часов (GC-MS control). Затем реакционная масса была разбавлена Et2O, промыта водой и солевым раствором. Органический слой осушали MgSO4 и и концентрировали при пониженном давлении Остаток очищали на хроматографической колонке с силикагелем (hexane:EtOAc, 97:3) получая продукт 3.
1,1,2,2-Тетрафтор-4-иодододекан (3a): Выход: 80%; бесцветное масло, 1H ЯМР (400 MHz, CDCl3) δ 5.72 (tdd, J = 53.9, 3.0, 2.3 Hz, 1H), 4.39–4.27 (m, 1H), 2.96–2.57 (m, 2H), 1.91–1.69 (m, 2H), 1.61–1.38 (m, 2H), 1.36–1.21 (m, 10H), 0.89 (t, J = 7.1 Hz, 3H); 13C ЯМР (101 MHz, CDCl3) δ 117.52 (tt, J = 249.2, 29.4 Hz), 110.03 (tt, J = 249.9, 41.1 Hz), 40.85 (t, J = 21.2 Hz), 40.64, 31.99, 29.73, 29.51, 29.37, 28.71, 22.81, 22.41 (t, J = 2.0 Hz), 14.24; 19F ЯМР (377 MHz, CDCl3) δ –112.75 to –117.32 (m, 2F), –134.35 to –137.52 (m, 2F).; HRMS (EI+) m/z вычислено для C12H21F4I [M]+: 368.0624, найдено 368.0621.
(5,5,6,6- Тетрафтор -3-иодогексилбензол (3b): Выход: 92%; бесцветное масло, 1H ЯМР (400 MHz, CDCl3) δ 7.35–7.26 (m, 2H), 7.27–7.17 (m, 3H), 5.70 (tdd, J = 53.9, 3.0, 2.2 Hz, 1H), 4.28 (tt, J = 8.4, 5.3 Hz, 1H), 3.00–2.63 (m, 4H), 2.13 (tt, J = 7.9, 5.0 Hz, 2H); 13C ЯМР (101 MHz, CDCl3) δ 140.25, 128.71, 128.66, 126.46, 117.50 (tt, J = 249.5, 29.6 Hz), 109.97 (tt, J = 250.1, 41.1 Hz), 42.06 (d, J = 2.1 Hz), 40.85 (t, J = 21.2 Hz), 35.84, 21.52 (t, J = 2.5 Hz).; 19F ЯМР (376 MHz, CDCl3) δ –111.96 to –116.66 (m, 2F), –133.82 to –136.55 (m, 2F); HRMS (EI+) m/z вычислено для C12H13F4I [M]+: 359.9998, найдено 359.9994.
12,12,13,13- Тетрафтор -10-иодотридекан-1-ол (3c): Выход: 88%; бесцветное масло, 1H ЯМР (400 MHz, CDCl3) δ 5.71 (tt, J = 53.9, 2.7 Hz, 1H), 4.31 (tt, J = 8.6, 5.1 Hz, 1H), 3.61 (t, J = 6.2 Hz, 2H), 2.89–2.58 (m, 2H), 2.02 (s, 1H), 1.84–1.70 (m, 2H), 1.59–1.49 (m, 2H), 1.40–1.21 (m, 12H); 13C ЯМР (101 MHz, CDCl3) δ 117.46 (tt, J = 249.1, 29.4 Hz), 109.96 (tt, J = 250.0, 41.0 Hz), 63.07, 40.78 (t, J = 21.1 Hz), 40.53 (d, J = 1.9 Hz), 32.86, 29.64, 29.57, 29.46, 29.40, 28.60, 25.82, 22.39; 19F ЯМР (376 MHz, CDCl3) δ –113.20 to –117.20 (m), –134.18 to –137.30 (m); HRMS (ESI+) m/z вычислено для C13H24F4IO [M+H]+: 399.0808, найдено 399.0811.
7,7,8,8- Тетрафтор -5-иодооктан-2-он (3d): Выход: 95%; бесцветное масло, 1H ЯМР (400 MHz, CDCl3) δ 5.74 (tdd, J = 53.8, 3.2, 2.2 Hz, 1H), 4.49–4.23 (m, 1H), 2.99–2.55 (m, 4H), 2.17 (s, 3H), 2.16–2.07 (m, 1H), 1.97 (dddd, J = 15.2, 9.8, 8.1, 5.4 Hz, 1H); 13C ЯМР (101 MHz, CDCl3) δ 206.79, 117.34 (tt, J = 249.4, 29.4 Hz), 109.92 (tt, J = 250.1, 40.8 Hz), 43.75, 41.03 (t, J = 21.3 Hz), 34.25 (d, J = 2.2 Hz), 30.21, 21.21 (t, J = 2.7 Hz); 19F ЯМР (376 MHz, CDCl3) δ –112.26 to –116.45 (m), –134.10 to –136.77 (m); HRMS (ESI+) m/z вычислено для C8H12F4IO [M+H]+: 326.9869, найдено 326.9866.
13-Бром-1,1,2,2-тетрафтор-4-иодотридекан (3e): Выход: 83%; бесцветное масло, 1H ЯМР (400 MHz, CDCl3) δ 5.72 (tdd, J = 53.9, 3.1, 2.2 Hz, 1H), 4.32 (tdd, J = 8.3, 5.8, 4.3 Hz, 1H), 3.40 (t, J = 6.8 Hz, 2H), 2.92–2.55 (m, 2H), 1.90–1.80 (m, 4H), 1.36–1.23 (m, 12H); 13C ЯМР (101 MHz, CDCl3) δ 117.48 (tt, J = 249.1, 29.4 Hz), 114.26, 109.98 (tt, J = 250.0, 41.0 Hz), 40.81 (t, J = 21.2 Hz), 40.54 (d, J = 1.9 Hz), 34.11, 32.93, 29.65, 29.42, 29.37, 28.81, 28.59, 28.26, 22.37; 19F ЯМР (376 MHz, CDCl3) δ –112.92 to –117.43 (m), –134.50 to –137.09 (m); HRMS (EI+) m/z вычислено для C13H21F4BrI [M–H]+: 458.9808, найдено 368.9799.
12,12,13,13- Тетрафтор -10-иодотридекановая кислота (3f): Выход: 97%; бесцветное масло, 1H ЯМР (400 MHz, CDCl3) δ 5.72 (tt, J = 53.9, 2.7 Hz, 1H), 4.32 (tdd, J = 8.3, 5.7, 4.3 Hz, 1H), 2.95–2.55 (m, 2H), 2.35 (t, J = 7.5 Hz, 2H), 1.79 (ddp, J = 19.7, 9.8, 4.5 Hz, 2H), 1.63 (p, J = 7.4 Hz, 2H), 1.53 (tq, J = 7.7, 3.8, 2.8 Hz, 1H), 1.42–1.29 (m, 9H); 13C ЯМР (101 MHz, CDCl3) δ 180.37, 117.51 (tt, J = 249.1, 29.4 Hz), 110.01 (tt, J = 250.1, 41.1 Hz), 40.81 (t, J = 21.1 Hz), 40.54 (d, J = 1.9 Hz), 34.18, 29.65, 29.27, 29.24, 29.10, 28.57, 24.75, 22.37 (t, J = 2.5 Hz); 19F ЯМР (376 MHz, CDCl3) δ –112.70 to –117.55 (m), –134.26 to –137.53 (m); HRMS (ESI+) m/z вычислено для C13H22F4IO2 [M+H]+: 413.0601, найдено 413.0602.
2-(9,9,10,10- Тетрафтор -7иододецил)оксиран (3g): Выход: 74%; бесцветное масло, 1H ЯМР (400 MHz, CDCl3) δ 5.71 (tdd, J = 53.9, 3.2, 2.2 Hz, 1H), 4.31 (tdd, J = 8.3, 5.7, 4.3 Hz, 1H), 2.89 (tdd, J = 5.0, 3.9, 2.7 Hz, 1H), 2.86–2.60 (m, 2H), 2.77–2.68 (m, 1H), 2.45 (dd, J = 5.0, 2.7 Hz, 1H), 1.78 (ddt, J = 14.7, 9.6, 5.0 Hz, 2H), 1.58–1.22 (m, 10H); 13C ЯМР (101 MHz, CDCl3) δ 117.48 (tt, J = 249.2, 29.4 Hz), 109.97 (tt, J = 250.1, 41.0 Hz), 52.41, 47.18, 40.79 (t, J = 21.2 Hz), 40.46 (d, J = 1.8 Hz), 32.52, 29.57, 29.28, 28.54 (d, J = 1.8 Hz), 25.99, 22.27 (t, J = 2.5 Hz); 19F ЯМР (376 MHz, CDCl3) δ –112.73 to –117.45 (m), –134.39 to –137.12 (m); HRMS (ESI+) m/z вычислено для C12H20F4IO [M+H]+: 383.0495, найдено 383.0493.
Синтез соединения 5 (диэтил цис-3-(иодометил)-4-(2,2,3,3-тетрафторпропил)циклопентан -1,1-дикарбоксилата:
Диэтил 2,2-диаллилмалонат (4) (120 mg, 0.5 mmol) и вода (5 mL) охлаждали до 0 °C и дегазировали трехкратным заполнением с помощью насоса атмосферой азота. К этой эмульсии добавляли Rhodamine B (12 mg, 0.025 mmol) и затем иодид 2 (227 mg, 103 μL, 1.0 mmol) с помощью предварительно охлажденного до 0 °C дозатора (шприца). Перемешиваемая реакционная смесь была нагрета до комнатной температуры и облучалась синей LED лампой в течении 16 часов (GC-MS control). Затем реакционная масса была разбавлена Et2O, промыта водой и солевым раствором. Органический слой осушали MgSO4 и концентрировали при пониженном давлении Остаток очищали на хроматографической колонке с силикагелем (hexane:EtOAc, 99:1) получая продукт 5 (180 mg желтого масла, 76% Выход) как смесь цис:транс изомеров в соотношении 95:5. 1H ЯМР (400 MHz, CDCl3) δ 5.70 (tt, J = 54.0, 2.6 Hz, 1H), 5.11–5.07 (m, 1H), 5.06 (t, J = 1.2 Hz, 1H), 4.26–4.08 (m, 4H), 3.18 (ddd, J = 9.8, 5.2, 1.0 Hz, 1H), 3.01 (t, J = 9.9 Hz, 1H), 2.61 (dt, J = 7.5, 1.2 Hz, 2H), 2.57–2.43 (m, 2H), 2.34–2.18 (m, 1H), 2.14–1.82 (m, 1H), 1.28–1.15 (m, 6H); 13C ЯМР (101 MHz, CDCl3) δ 172.03, 170.70, 132.30, 119.09, 117.85 (tt, J = 247.3, 29.5 Hz), 110.11 (tt, J = 249.4, 41.6 Hz), 61.76 (d, J = 9.6 Hz), 61.19, 45.44, 39.73, 36.70, 35.41 (d, J = 2.2 Hz), 28.45 (t, J = 21.8 Hz), 14.09, 13.97 (d, J = 3.0 Hz); 19F ЯМР (376 MHz, CDCl3) δ –102.18 to –108.83 (m), –125.64 (ddd, J = 53.3, 38.9, 8.5 Hz); HRMS (ESI+) m/z вычислено для C15H22F4IO4 [M+H]+: 469.04934, найдено 469.04935.
Добавление HCF2CF2I к диэтил 2-аллилмалонату 6:
а) Реакция катализирована Rhodamine B. Смесь диэтил 2-аллилмалоната (6) (200 mg, 1 mmol) и воды (10 mL) охлаждали до 0 °C и дегазировали трехкратным заполнением с помощью насоса атмосферой азота. К этой эмульсии добавляли (i-Pr)2NEt (192 μL, 1.10 mmol), Rhodamine B (24 mg, 0.050 mmol), и HCF2CF2I (227 mg, 103 μL, 1.0 mmol) с помощью предварительно охлажденного до 0 °C дозатора (шприца). Перемешиваемая реакционная смесь была нагрета до комнатной температуры и облучалась синей LED лампой в течении 16 часов (GC-MS control).
Реакционнная масса по данным GC-MS анализа содержала смесь продукта присоединения-отщепления 7a и продукта присоединения 7b.
Темно-красная суспензия была разбавлена Et2O, промыта водой и солевым раствором. Органический слой осушали MgSO4 и концентрировали при пониженном давлении. Остаток очищали на хроматографической колонке с силикагелем (hexane:EtOAc, 95:5) получая смесь 7a и 7b (250 mg темно-желтого масла, 58% Выход, 7a:7b 4:1)
б) Реакция катализирована Eosin Y.
Смесь диэтил 2-аллилмалоната (6) (200 mg, 1 mmol) и воды (10 mL) охлаждали до 0 °C и дегазировали трехкратным заполнением с помощью насоса атмосферой азота.К этой эмульсии добавляли Cs2CO3 (652 mg, 2 mmol), Eosin Y (32 mg, 0.05 mmol), и HCF2CF2I (454 mg, 206 μL, 2.0 mmol) ) с помощью предварительно охлажденного до 0 °C дозатора (шприца). Перемешиваемая реакционная смесь была нагрета до комнатной температуры и облучалась синей LED лампой в течении 20 часов. Реакционная смесь по данным GC-MS была чистым продуктом присоединения-отщепления 7a . Красная суспензия была разбавлена Et2O, промыта водой и солевым раствором.
Органический слой осушали MgSO4 и концентрировали при пониженном давлении.
Выход 247 mg (82%) бесцветное масло 7a. Диэтил(E)-2-(4,4,5,5тетрафторпент-2-ен-1-ил)малонат (7a): 1H ЯМР (400 MHz, CDCl3) δ 5.75 (tt, J = 53.8, 2.9 Hz, 1H), 4.34–4.07 (m, 4H), 2.40–2.21 (m, 1H), 2.15–2.00 (m, 1H), 1.97–1.77 (m, 1H), 1.54–1.43 (m, 2H), 1.26 (td, J = 7.1, 5.5 Hz, 6H); 13C ЯМР (101 MHz, CDCl3) δ 169.47, 167.89, 117.38 (tt, J = 247.3, 29.4 Hz), 110.11 (tt, J = 249.4, 40.3 Hz), 61.87, 61.85, 32.99, 29.12 (t, J = 22.4 Hz), 20.25, 19.62 (t, J = 5.5 Hz), 14.15, 14.09; 19F ЯМР (376 MHz, CDCl3) δ –116.19 (t, J = 17.2 Hz), –136.03 (dd, J = 53.7, 26.3 Hz); HRMS (ESI+) m/z вычислено для C12H17F4O4 [M+H]+: 301.1063, найдено 301.1062.
Восстановление (5,5,6,6-тетрафтор-3-иодогексил)бензола (3b). (5,5,6,6-Тетрафторгексил)бензол (8):
Соединение 3b (150 mg, 0.42 mmol) растворяли в безводном тетрагидрофуране (2 mL) и охлаждали до 0 °C. Затем добавляли LiAlH4 (30 mg, 0.79 mmol) и реакционной смеси позволяли нагреватьсяя до комнатной температуры. При перемешиваниии в течении 16 часов (GC-MS control). Насыщенный раствор NH4Cl (2 mL) и воды (30 mL) были добавлены и реакционная масса экстрагировалась Et2O. Органический слой осушали MgSO4 и концентрировали при пониженном давлении. Остаток был очищен на хроматографической колонке с силикагелем с помощью гексана, давая продукт 8 (75 mg бесцветное масло, 77% Выход). 1H ЯМР (400 MHz, CDCl3) δ 7.34–7.29 (m, 2H), 7.25–7.18 (m, 3H), 5.71 (tt, J = 54.1, 2.8 Hz, 1H), 2.67 (t, J = 7.5 Hz, 2H), 2.10–1.91 (m, 2H), 1.78–1.69 (m, 2H), 1.69–1.60 (m, 2H); 13C ЯМР (101 MHz, CDCl3) δ 141.99, 128.53, 128.50, 126.04, 118.13 (tt, J = 245.8, 29.2 Hz), 110.47 (tt, J = 249.1, 41.5 Hz), 35.73, 31.22, 29.78 (t, J = 22.5 Hz), 20.20 (t, J = 3.8 Hz); 19F ЯМР (376 MHz, CDCl3) δ –116.11 (td, J = 18.2, 2.0 Hz), –135.45 (d, J = 54.0 Hz); HRMS (EI+) m/z вычислено для C12H14F4 [M]+: 234.1032, найдено 234.1033.
Благодарности: Данная работа была финансово поддержана Академией наук Чехии (RVO: 61388963). Мы также благодарны P&M Invest Ltd. за бесплатные образцы HCF2CF2I.
References
- Dolbier, W. R. Chem. Rev. 1996, 96, 1557.
- Studer, A. Angew. Chem., Int. Ed. 2012, 51, 8950.
- Ji, Y.; Brueckl, T.; Baxter, R. D.; Fujiwara, Y.; Seiple, I. B.; Su, S.; Blackmond, D. G.; Baran, P. S. Proc. Natl. Acad. Sci. USA 2011, 108, 14411.
- Wei-Yuan, H.; Yuan, X. Chin. J. Chem. 1990, 8, 362.
- Fujiwara, Y.; Dixon, J. A.; O'Hara, F.; Funder, E. D.; Dixon, D. D.; Rodriguez, R. A.; Baxter, R. D.; Herle, B.; Sach, N.; Collins, M. R.; Ishihara, Y.; Baran, P. S. Nature 2012, 492, 95.
- Zhang, C. Adv. Synth. Cat. 2014, 356, 2895.
- Nagib, D. A.; MacMillan, D. W. Nature 2011, 480, 224.
- Greiner, J.; Milius, A.; Riess, J. G. J. Fluorine Chem. 1992, 56, 285.
- Takeyama, Y.; Ichinose, Y.; Oshima, K.; Utimoto, K. Tetrahedron Lett. 1989, 30, 3159.
- Bravo, A.; Bjørsvik, H.-R.; Fontana, F.; Liguori, L.; Mele, A.; Minisci, F. J. Org. Chem. 1997, 62, 7128.
- Chen, Q.-Y.; Qiu, Z.-M.; Yang, Z.-Y. J. Fluorine Chem. 1987, 36, 149.
- Beier, P.; O’Hagan, D.; Pearson, C.; Petty, M. C.; Slawin, A. M. Z. J. Fluorine Chem. 2005, 126, 671.
- Zhang, C. P.; Chen, Q. Y.; Guo, Y.; Xiao, J. C.; Gu, Y. C. Chem. Soc. Rev. 2012, 41, 4536.
- Huang, W.-Y.; Zhang, H.-Z. J. Fluorine Chem. 1990, 50, 133.
- Habib, M. H.; Mallouk, T. E. J. Fluorine Chem. 1991, 53, 53.
- Qiu, Z.-M.; Burton, D. J. J. Org. Chem. 1995, 60, 3465.
- Mizuta, S.; Verhoog, S.; Engle, K. M.; Khotavivattana, T.; O'Duill, M.; Wheelhouse, K.; Rassias, G.; Medebielle, M.; Gouverneur, V. J. Am. Chem. Soc. 2013, 135, 2505.
- Iqbal, N.; Jung, J.; Park, S.; Cho, E. J. Angew. Chem., Int. Ed. Engl. 2014, 53, 539.
- Wozniak, L.; Murphy, J. J.; Melchiorre, P. J. Am. Chem. Soc. 2015, 137, 5678.
- Wang, Y.; Wang, J.; Li, G. X.; He, G.; Chen, G. Org. Lett. 2017, 19, 1442.
- Chernykh, Y.; Hlat-Glembová, K.; Klepetářová, B.; Beier, P. Eur. J. Org. Chem. 2011, 2011, 4528.
- Beier, P.; Chernykh, Y.; Opekar, S.; Klepetářová, B. Synlett 2012, 23, 1187.
- Chernykh, Y.; Beier, P. J. Fluorine Chem. 2013, 156, 307.
- Chernykh, Y.; Jurásek, B.; Beier, P. J. Fluorine Chem. 2015, 171, 162.
- Václavík, J.; Chernykh, Y.; Jurásek, B.; Beier, P. J. Fluorine Chem. 2015, 169, 24.
- Li, L.; Ni, C.; Xie, Q.; Hu, M.; Wang, F.; Hu, J. Angew. Chem., Int. Ed. Engl. 2017, 56, 9971.
- Yoshioka, E.; Kohtani, S.; Jichu, T.; Fukazawa, T.; Nagai, T.; Kawashima, A.; Takemoto, Y.; Miyabe, H. J. Org. Chem. 2016, 81, 7217.
- Tiwari, D. P.; Dabral, S.; Wen, J.; Wiesenthal, J.; Terhorst, S.; Bolm, C. Org. Lett. 2017, 19, 4295.
Статью рекомендовал к публикации член редколлегии С. М. Игумнов
Fluorine Notes, 2017, 115, 3-4


