Поступило в редакцию: Сентябрь 2017
УДК 10.17677/fn20714807.2017.05.04
Fluorine Notes, 2017, 114, 7-8
Синтез 2,2’-азанедиил-бис((N,N-бис(4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-гептадекафторандецил) ацетамида) – нового фторсодержащего вторичного амина с четырьмя звеньями перфтороктила
Anikó Nemes, Gitta Schlosser, Antal Csámpai, Dénes Szabó and József Rábai*
Institute of Chemistry, Eötvös Loránd University, Pázmány Péter sétány 1-A, 1117 Budapest, Hungary
E-mail: rabai@elte.hu
Аннотация: Бис(перфтороктилпропил)амин и ди(пентафторфенил) N-(бензилоксикарбонил)иминацетат были нагреты в C6H5CF3 образуя фторированные диамиды с четырьмя перфтороктиловыми звеньями, которые со снятием защиты при каталитическом гидрировании в растворителе FC-72 (смесь перфторгексанов) давали соответствующие вторичные амины с хорошим выходом. Несмотря на то, что фторированные вторичные амины показали довольно низкую растворимость в перфторалканах при нормальных условиях, было установлено их роль в повышении растворимости коллоидных частиц платины в эфире, бензотрифториде и перфторалканах.
Ключевые слова: фторсодержащие амины, фторные акцепторы, фторные дендримеры
Фторированные первичные (1o), вторичные (2o) и третичные (3o) амины типа [Rfn(CH2)mRfn]xNH3-x) являются важными конструкционными блоками и реагентами в химии фтора [1]. Их прекурсоры фторированных альдегидов и фторалкил иодидов достаточно доступны [2]. Вторичный амин [(C6F13CH2CH2)3SiCH2CH2CH2]2NH с шестью перфторгексилами был синтезирован и применен как фтор-акцептор мочевины в автоматическом параллельном синтезе в жидкой фазе, осуществленном Curran с сотр. [3]. Другое применение сильной фторной поддержки в виде шести перфтороктильных звеньев (Схема 1) позволяло провести синтез биоактивного пептида Лей-энкефалина с использованием Fmoc-стратегии как сообщал Mizuno с сотр. [4].

Схема 1. Структура с сильной фторной поддержкой, используемая для пептидного синтеза [4].
Синтез нового фторичного фторсодержащего амина с четырьмя перфтороктильными группами (6) описан здесь, который включает реакцию бис(перфтороктил пропил)амина (4) и ди-пентафторфенилового эфира N-(бензилоксикарбонил)иминодиуксусной кислоты (3) с последующей депротекцией промежуточного фторного карбамата (5) при использовании каталитического гидрирования в растворителе FC-72 [5] (Схема 2).
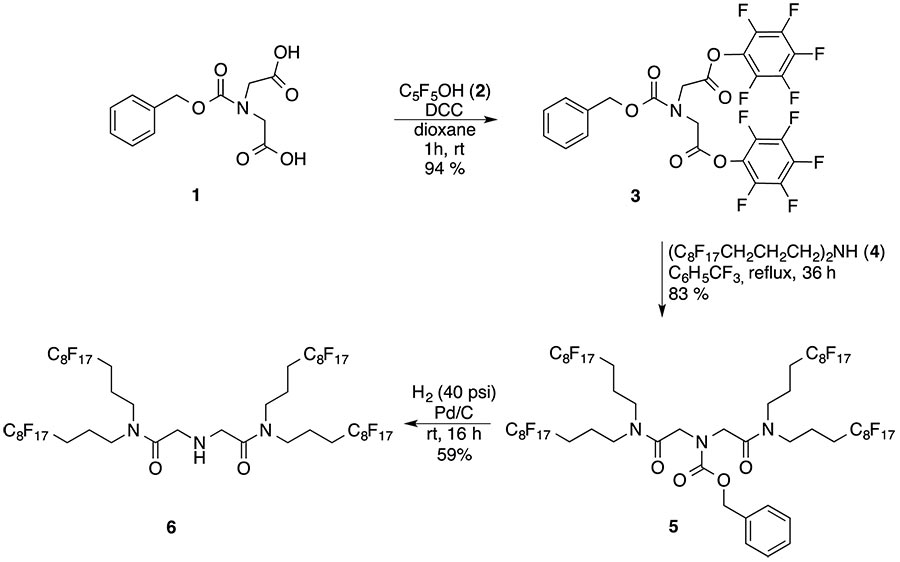
Схема 2. Синтез фторного амина 6 дендримерного типа c четыремя перфтороктильными звеньями.
Впрочем, синтез прекурсура активного эфира 3 был оптимизирован, давая выход 94%, применением перекристаллизации N-защищенной имидодиуксусной кислоты вместе с свежеперегнанным пентафторфенолом и исключением воздействия влаги воздуха. Реакция 3 (1 mol) и амина 4 (2 mol) имела место при гомогенных условиях в бензорифториде, давая 5-защищенный диамид с четырьмя перфтороктильными группами с 83% выходом, который представлял собой палевое желтое масло после двухфазного экстрактивного выделения.
Карбамат 5 (%F = 61.35) показал высокую растворимость в перфторалканах, тогда как твердый фторсодержащий амин 6 (%F= 65.52), полученный каталитическим гидрированием 5 на Pd/C, показал относительно низкую растворимость во фторсодержащих растворителях и имел Тпл. 99-101oC.
Это поведение находится в соответствии с эмпирическими принципами, которые предсказывают, что внутримолекулярное аттрактивное взаимодействие должно быть минимизировано, для того чтобы достичь высокого сродства к фтору и высокой растворимости в фторуглеродах [6].
Поэтому амин 6, несмотря на более высокое содержание фтора, чем в прекурсоре карбамате 5, не подходит по требованиям для применения как акцептор фтора [7]. Однако, соединение 6 может использоваться как фторсодержащий конструкционный блок или как реагент для стабилизации и придания растворимости коллоидов Pd в перфторалканах. Небольшое количество смеси выпавшего в осадок 6 и Pd/C были нагреты с несколькими миллилитрами перфторметилциклогексана (CF3C6F11) в течении минуты и затем отфильтрованы, давая ярко-коричневый раствор. Это наблюдение является индикацией образования растворимого во фторуглероде палладиевого коллоида. После того, как этот раствор встряхивали с Ph3P в толуоле, получали бесцветный слой перфторметилциклогексана, но вскоре на границе фаз появлялся черный осадок. В различных экспериментах такие коллоиды палладия разлагали полностью кипячением в течении нескольких минут.
Экспериментальная часть
Прекурсоры 1 и 4 были получены как сообщено в работах [8] и [1b] соответственно. Фторированная инертная жидкость FC-72, бензотрифторид и CF3CH2OH были приобретены у FC Chemicals, тогда как другие реагенты и органические растворители были приобретены у Sigma-Aldrich и MOLAR. 1H-, 13C- и 19F-ЯМР спектры были получены на приборе Bruker Avance 250 с использованием инверсионной 5 мм 1H/13C/31P/19F измерительной головки при комнатной температуре. Химические сдвиги (δ) даны в м.д. относительно остаточных пиков растворителя (CDCl3) (δ=7.26 дляr 1H, δ=77.0 для 13C) и к CFCl3 как внешнему стандарту (δ=0.00 для 19F). Определение молекулярной массы и сбор данных тандемных масс-спектров проводили масс-спектрометрией с ионизацией распылением в электрическом поле (ESI-MS) на масс-спектрометре Bruker Daltonics Esquire 3000 plus (Германия) с ионной ловушкой. Образцы растворяли в смеси растворителей ацетонитрил-трифторэтанол (50:50 об.) Масс спектры были получены в диапазоне 200-3000 m/z. Напряжение на капилляре было 4000 В, давление газа в капилляре 10 psi (фунтов на квадратный дюйм), расход осушающего газа (drying gas flow) – 4 л в мин., температура капилляра 250 oC. Образцы вводили в источник ионов в потоке 10 L/мин. используя поршневой насос.
Температуры плавления определяли с помощью аппарата Боеция для микро-определения точки плавления и не корректировали. ГЖХ анализ летучих продуктов определяли на приборе Hewlett-Packard 5890 Series II с PONA [сшитая метилсиликоновая смола] 50 m x 0.2mm x 0.5 mm колонкой, H2 газ-носитель, детектор ДИП; Программа: 120 °C, 5 мин., 10 °C/мин., 250 °C, 5 мин., Температура инжектора: 250°C, Детектора: 280°C.
Бис(пентафторфенил) 2,2 '(((бензилокси)карбонил)азанедиил)диацетат (3)
В осушенную прокаливанием колбу и в атмосфере аргона при перемешивании магнитной мешалкой смеси N-(бензилоксикарбонил)-имнодиуксусной кислоты (1) (3.00g, 11.2 mmol) and C6F5OH (2) (4.58 g, 24.6 mmol) в абсолютном диоксане (60 mL) был по каплям добавлен раствор 1,3-дициклогексилкарбодиимида (DCC) абсолютном диоксане (12 mL) при комнатной температуре в течении 1 часа. Прогресс реакции наблюдали по образованию густого белого осадка (DCU, N,N′-Dicyclohexylurea, N,N'-дициклогексилмочевина). Смесь оставляли при комнатной температуре на ночь, затем осадок отфильтровывали.
Осадок промывали диоксаном (3 × 10 mL) и сушили с получением 5.05 g (22.6 mmol, 92%) DCU как побочного продукта. Фильтрат испаряли используя роторный испаритель при температуре бани 50 oC и остаточном давлении 16 мм. рт. ст. в течении 30 минут с получением 8.2 г расплавленного «сырца», который затвердевал при охлаждении до комнатной температуры. Перекристаллизация из н-гептана (50 mL) дала 5.10 g (76%) белого кристаллического продукта с Тпл.=80-82oC. Масштабирование этого синтеза в 4 раза (т.е. 12.0 g соединения 1 [C6H5CH2OC(O)N(CH2CO2H)2]) привело к получению 25.2 g (94%) продукта той же самой чистоты, как и ранее. Спектральные характеристики согласуются с сообщенными в работе [9].
1H-ЯМР (CDCl3) (, м.д.): 7.29, s, (5H, C6H5); 5.17, s, (2H, PhCH2); 4.46, s, (NCH2C=O, A-цепь); 4.55, s, (NCH2C=O, B- цепь).
13C-ЯМР (CDCl3) (, м.д.): 128.9 (Car-4); 129.0 (Car-3,5); 128.5 (Car-2,6); 135.7 (Car-1); 69.3 (CH2OCO); 155.8 (CH2OCO); 49.1 (CH2CO2, А-цепь); 165.9 (CH2CO2, А-цепь); 49.4 (CH2CO2, В-цепь); 166.0 (CH2CO2, В-цепь).
Бензил бис(2-(бис(4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-гептадекафторандецил)амино)-2-оксоэтил) карбамат (5)
В атмосфере аргона перемешиваемая смесь 4 (7.12 g, 7.60 mmol) и активного эфира 3 (2.28 g, 3.80 mmol) в C6H5CF3 (30 mL) нагревалась в колбе с обратным холодильником при температуре масляной бани 130oC до тех пор, пока хроматографический анализ образца не показал, что боле не происходит увеличение соотношения 2:4. Это заняло примерно 36 часов. С помощью роторного испарителя растворитель испаряли, после чего остающуюся масляную жидкость растворяли в FC-72 (35 mL) – CH3OH би-фазной системе. Побочный C6F5OH преимущественно был в CH3OH фазе. Нижняя фтористая фаза была отделена и последовательно промыта CH3OH (2 × 15 mL) и 2M NaOH (2 × 50 mL). Разделение фаз, которое показало четкий верхний водный слой, было облегчено добавлением нескольких капель CH3OH. Слой FC-72 был промыт снова CH3OH (2 × 15 mL), после чего был отделен и осушен (Na2SO4). Фильтрат концентрировали отгонкой растворителя при атмосферном давлении, что позволило регенерировать большую часть растворителя FC-72 (25 mL). Следы FC-72 из оставшейся масляной жидкости были удалены при 16 мм. рт. ст. и температуре 110oC в течении 1 часа. Выход: 6.65 g (83%), бледно-жёлтое масло. Соединение 5 полностью смешивается с перфторалканами, бензотрифторидом и эфиром. Соединения 2 и 4, как определено хроматографией, отсутствовали.
1H-ЯМР (CF2ClCFCl2-CDCl3, 1:1 v/v, TMS) (, м.д.): 4.17, s, (2H, NCH2CO-, А-цепь); 4.25 s, (2H, NCH2CO-, В-цепь); 5.04 s, (2H, PhCH2OCO-); 7.13,~s, (5H, C6H5CH2).
13C-ЯМР(CF2ClCFCl2-CDCl3, 1:1 v/v, TMS) (, м.д.): 136.3 (Car-1); 128.5 (Car-2, 6); 128.7 (Car-3,5); 128.6 (Car-4); 68.5 (PhCH2); 156.4 (PhCH2OC=O); 48.7 (CH2C(=O)N, А-цепь); 168.6 (CH2C(=O)N, А-цепь); 47.1 (NCH2CH2, A-1-chain); 45.7 (NCH2CH2, A-2-chain); 49.4 (CH2C(=O)N, В-цепь); 169.1 (CH2C(=O)N, В-цепь); 46.7 (NCH2CH2, B-1-chain); 45.8 (NCH2CH2, B-2-chain).
2,2’-азанедиил-бис((N,N-бис(4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-гептадекафторандецил) ацетамид) (6)
В 250 мл стеклянный сосуд автоклава среднего давления поместили карбамат 5 (6.74 g, 3.2 mmol), растворенный в FC-72 (60 mL), и 10% Pd/C катализатора (600 mg Pd-C). После этого смесь гидрировали при давлении водорода 40 psi (фунтов на квадратный дюйм) при комнатной температуре в течении 16 часов. Сырец был выделен фильтрацией, промыт FC-72 и осушен под вакуумом. Выход: 6.27 g (~ 90%) беловатого твердого вещества (6 и Pd/C). Этот сырец экстрагировали кипящим эфиром в аппарате Сокслета в течении 48 часов. Экстракт испаряли в роторном испарителе и затем полученный кристаллический продукт отфильтровывали, промывали CH3OH и сушили над P2O5 в вакууме. Выход: 3.69 g (59%), белый кристаллический продук, Тпл. = 99-101oC.
1H-ЯМР (CF2ClCFCl2-CDCl3, 1:1 v/v, TMS) (, м.д.): 3.51, s, (2H, NCH2CO); 3.45, t, (2H, 7.6 Hz, NCH2CH2, А-цепь); 3.37, t, (2H, 7.6 Hz, NCH2CH2, В-цепь); 2.12, br, (4H, NCH2CH2, А-цепь и В-цепь); 1.92, tt, (4H, 14.4 Hz, 7.3 Hz, А-цепь and В-цепь).
13C-ЯМР: 50.5 (H-NCH2); 171.2 (C=O); 45.6 (N-CH2CH2CH2, А-цепь); 20.6 (N-CH2CH2CH2, А-цепь); 29.1, t, (N-CH2CH2CH2C8F17, А-цепь); 46.9 (N-CH2CH2CH2, В-цепь); 19.5 (N-CH2CH2CH2, В-цепь); 28.7, t, (N-CH2CH2CH2C8F17, В-цепь).
MS(ESI) (CH3CN : CF3CH2OH = 1:1 v/v): Вычислено:
C48H29F68N3O2 = 1971.1; Получено: 1971.1
Благодарности
Мы благодарим National Research, Development and Innovation Office за финансовую поддержкуe M-ERA.Net COR_ID программы (NKFIH NN117633). G. S. благодарит за поддержку MTA János Bolyai Research Scholarship и MTA Premium Post-Doctorate Research Program Венгерской Академии наук (HAS, MTA).
Дополнительная информация
На Рис. 1 приведен масс спектр соединения 6.
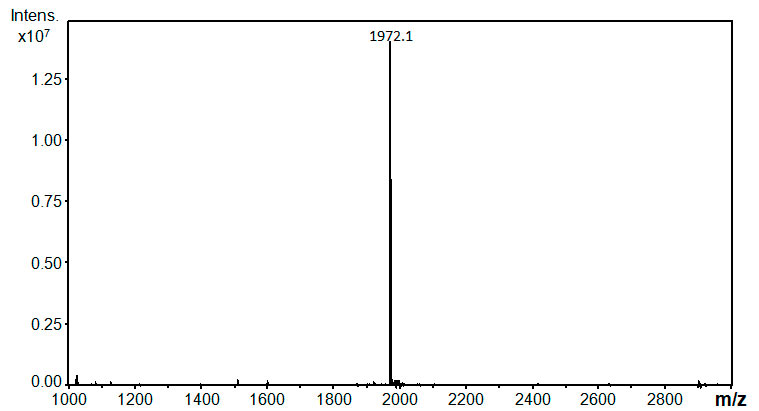
Рисунок 1. ESI-MS спектр соединения 6 на масс-спектрометре Bruker Daltonics Esquire 3000 plus (Германия) с ионной ловушкой. Образец был растворен в смеси ацетонитрил- трифторэтанол (50:50 об.) Зарегистрированный ион соответствует протонированной молекуле [M+H]+.
Литература
- (a) C. Rocaboy, W. Bauer, and J.A. Gladysz, Convenient Syntheses of a Family
of Easily Recoverable Fluorous Primary, Secondary, and Tertiary Aliphatic Amines NH3-x[(CH2)m(CF2)7CF3]x(m=
3-5; x=1, 2, 3) - FineTuning of Basicities and Fluorous Phase Affinities, Eur. J. Org. Chem. 2000,
2621-2628;
DOI: 10.1002/1099-0690(200007)2000:14<2621::AID-EJOC2621>3.0.CO;2-H
(b) Z. Szlávik, G. Tárkányi, Á. Gömöry, Gy. Tarczay, J. Rábai,Convenient syntheses and characterization of fluorophilic perfluorooctyl-propylamines and ab initio calculations of proton affinities of related model compounds, J. Fluorine Chem.2001, 108, 7-14.
https://doi.org/10.1016/S0022-1139(00)00398-5
(c) Szabó, D.; Mohl, J.; Bálint, A.-M.; Bodor, A.; Rábai, J. Novel generation ponytails in fluorous chemistry: Synthesis of primary, secondary, and tertiary (nonafluoro-tert-butyloxy)ethyl amines. J. Fluorine Chem.2006,127, 1496-1504; DOI: 10.1016/j.jfluchem.2006.06.020 - (a) Nemes, A.; Berta, M.; Ivanko, P.; Szabó, D.; Rábai, J. Synthesis of 3-perfluoroalkylpropanals
and 3-perfluoroalkylpropionitriles. FLUORINE NOTES / FTORNIE ZAMETKI: 6(103):November-December (2015);
DOI 10.17677/fn20714807.2015.06.02;
(b) Menczinger, B.; Jakab, G.; Szabó, D.; Rábai, J. Synthesis of 1-iodo-3-perfluoroalkylpropanes and 1-iodo-4-perfluoroalkylbutanes. FLUORINE NOTES / FTORNIE ZAMETKI: (3) 94. May-June (2014). - Linclau, B., Singh, A.K. and Curran, D.P., Organic-fluorous phase switches: A fluorous amine scavenger for purification in solution phase parallel synthesis.J. Org. Chem.,1999, 64, 2835–2842. DOI:10.1021/jo9823442
- Mizuno, M.; Goto, K.;Miura, T.; Matsuura, T.; Inazu, T. Peptide synthesis on fluorous support.Tetrahedron Lett.2004, 45, 3425-3428. DOI: 10.1016/j.tetlet.2004.03.013
- http://multimedia.3m.com/mws/media/64892O/fluorinert-electronic-liquid-fc-72.pdf (accessed 03.08.2017)
- Kiss, L. E.; Kövesdi, I., Rábai, J. An Improved Design of FluorophilicMolecules: Prediction of the ln P Fluorous Partition Coefficient, Fluorophilicity, Using 3D QSAR Descriptors and Neural Networks. J. Fluorine Chem.2001,108,95-109.https://doi.org/10.1016/S0022-1139(01)00342-6
- (a) Lindsley, C. W.;Zhao, Z.; Leister, W. H.; Strauss, K. A. Fluorous-tethered amine bases for organic
and parallel synthesis: scope and limitations. Tetrahedron Letters,2002,
43, 6319-6323. https://doi.org/10.1016/S0040-4039(02)01399-0
(b) Chen, H.-T. C.; Zhang, W. Fluorous reagents and scavengers versus solid-supported reagents and scavengers, a reaction rate and kinetic comparision. Molecular Diversity, 2005, 9, 353-359. DOI: 10.1007/s11030-005-8104-3 - Abalain, C.; Langlois, M., Synthesis of dibenzyliminodiaceticacid derivatives as potential inhibitors of HIV-1 aspartyl protease. Eur. J. Med. Chem. Chim. Ther., 33 (2) 1998, 155-160. https://doi.org/10.1016/S0223-5234(98)80041-X
- (a) Lutter,
H.-D., Diederich, F. Synthesis of a macrobicyclic thiazolium-host and supramolecular catalysis of
the benzoin condensation. Angew. Chem., GE, 98 (12) 1986, 1125-1127; DOI: 10.1002/anie.198611251
(b) Diederich, F.; Schurmann, G.; Chao, I., Designed water-soluble macrocyclic esterases: from nonproductive to productive binding. J. Org. Chem.,53 (12), 1988, 2744-2757. DOI: 10.1021/jo00247a017
Статья рекомендована к публикации членом редколлегии József Rábai
Fluorine Notes, 2017, 114, 7-8


