Поступило в редакцию: Июль 2017
УДК 10.17677/fn20714807.2017.04.03
Fluorine Notes, 2017, 113, 5-6
Ассиметричная конструкция хиральных карбоновых центров с участием SCF3 фрагмента: прямое трифторметилтиолирование в сопоставлении с блочным принципом конструирования
Hélène Chachignon,1 Evgeniy V. Kondrashov,2 Dominique Cahard*,1
1 UMR 6014 CNRS C.O.B.R.A., Normandie Université, 1 rue Tesnière, 76821 Mont Saint Aignan, France
2 A. E. Favorsky Institute of Chemistry, Siberian Branch of the Russian Academy of Sciences, 1, Favorsky Str., Irkutsk 664033, Russia
Автор для корреспонденции: dominique.cahard@univ-rouen.fr
Аннотация: В данном мини-обзоре представлены недавние достижения в области ассиметричного синтеза хиральных фторсодержащих молекул содержащих SCF3 фрагмент. Рассмотрены как трифторметилтиолирование, так и использование фторированных фрагментов для конструирования молекул.
Ключевые слова: фтор – трифторметилтиолирование – ассиметричный синтез – хиральность
Содержание:
- Введение
- Прямое трифторметилтиолирование
- Диастереоселективное трифторметилтиолирование
- Энантиоселективное трифторметилтиолирование
- Реагенты для хирального трифторметилтиолирования
- Блочный принцип конструирования
- Перспективы
1. Введение
Фторорганическя химия является широчайшей областью исследования в связи с тем, что спектр применения фторированных молекул впечатляюще огромен, затрагивает все стороны общества от фармацевтики и агрохимии до материалов для науки и энергетики. В области наук о жизни роль хиральности особенно важна для взаимодействия молекул с биологической целью. Следовательно, стереоконтроль у карбоновых центров, содержащих фторированный фрагмент? является императивом (наиважнейшей задачей), чтобы продвигать на рынке энантиометрически чистые лекарств [1]. С точки зрения химии, ассиметрический синтез хиральных нерацемических фторированных веществ является крайне сложной задачей. Методы прямого введения атома фтора и трифторметильной группы многообразны и допускают высокий уровень стереоселективности [2]. С другой стороны, ассиметричное введение новых фторных фрагментов все еще находится в самом начале своего развития. Можно заметить, что из фторированных фрагментов, представляющих интерес в наше время, группа SCF3 стоит на первом месте по выбору [3]. На самом деле, исключительная липофильность, которую группа SCF3 передает молекулам (Hansch гидрофобный параметр: = 1.44 против 0.88 для CF3) и ее высокий электроноакцепторный показатель (константы Гаммета: σm = 0.40, σp = 0.50 против 0.43, 0.54, соответственно для CF3 группы) делают ее привлекательной для построения новейших химических архитектур для новых фармацевтических препаратов с улучшенной способностью проникать через клеточные мембраны. Большинство из настоящих трифторметиолированных биологически активных веществ имеют C(sp2)–SCF3 группу в основном в ароматической части, а два SCF3 фрагмента есть в препаратах, представленных на рынке для использования исключительно в ветеринарии: Toltrazuril или Baycox® от Bayer и Monepantel или Zolvix® от Novartis. Ни один из препаратов с SCF3 фрагментом не представлен на рынке в настоящий момент для лечения человека. Растущий интерес представляет развитие веществ, имеющих в составе C(sp3)–SCF3 группу, с фокусом на энантиомерно чистые молекулы. В данном контексте нам представляется своевременным сделать обзор синтетических подходов к ассиметричному конструированию хиральных карбоновых центров, несущих SCF3 фрагмент. Данный короткий, но в то же время исчерпывающий обзор содержит все материалы по этой теме, датированные до конца июня 2017.
2. Прямое трифторметилтиолирование
Прямое введение SCF3 группы в различные субстраты является наиболее распространенным подходом для конструирования хиральных карбоновых центров с SCF3 фрагментом. Среди применяемых стратегий большинство сфокусировано на энантиоселективном трифторметилтиолировании посредством хиральных катализаторов или хиральных реагентов. Диастереоселективное трифторметилтиолирование определенно не является разработанным методом с единичным опубликованным примером. Эти методы задействуют электрофильные реагенты трифторметилтиолирования, представленые на Рисунке 1 [4].
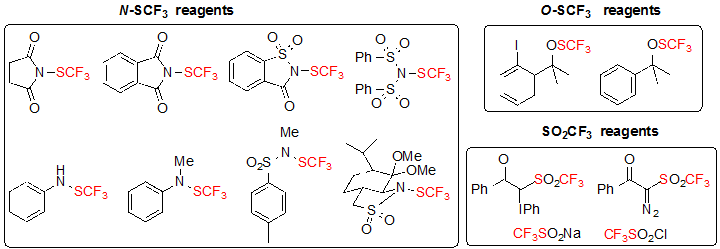
Рисунок 1. Классы электрофильных реагентов трифторметилтиолирования
2.1. Диастереоселективное трифторметилтиолирование
Шен и его коллеги впервые сообщили об ассиметричном введении SCF3 фрагмента в 2013. Эта группа предложила диастереоселективную трифторметилтиолирование литиевого енолята имеющнго хиральный фенилоксазолидин-2-он Эванса, используя трифторметилированный тиопероксид в качестве источника SCF3 (Схема 1) [5]. Ожидаемый продукт был получен с высокой диастереселективностью (dr 50:1), несмотря на низкий выход (20%). Однако, дальнейшие исследования условий реакции не проводились. В литературе до сегодняшнего дня также не сообщалось ни об одном другом примере.
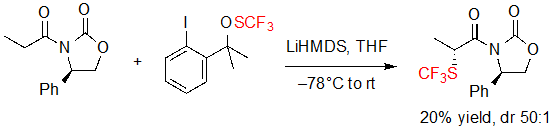
Схема 1. Диастереоселективноея трифторметилтиолирование карбонильного соединения.
2.2. Энантиоселективное трифторметилтиолирование
В противоположность к диастереоселективному трифторметилтиолированию, сообщений касающихся энантиоселективного введения SCF3 фрагментов на различных субстратах очень много.
Трифторметилтиолирование β-кетоэфиров
В 2013 Шен с коллегами описал первое ассиметричное органокаталитическое трифторметилтиолирование β-кетоэфиров с помощью катализаторов на основе хинина. Разнообразные инданон-производные адамантил β-кетоэфиров были легко превращены в свои (R)- трифторметилтиолированные производные с высокими выходами и превосходной энанатиоселективностью при применении 20 %(моль) хинина, а также 1.2 эквивалента реагента трифторметилсульфената Шена в толуоле при 40 °C (Схема 2) [6].
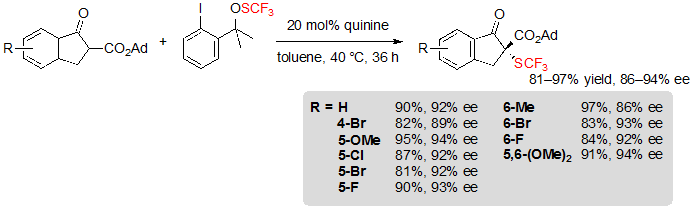
Схема 2. Ассиметричное трифторметилтиолирование адамантил β-кетоэфиров на основе инданона
Доказано, что энантиоселективность особенно чувствительна к стерической изоляции группы сложных эфиров; на самом же деле, показатели сильно падают при проведении реакции на метиловом или этиловом сложных эфирах. Напротив, природа и положение заместителей бензольного кольца на деле мало влияют на процесс реакции, так как такие же результаты были получены с субстратами, несущими электроноакцепторные или электронодонорные группы. Условия также показали хорошую совместимость с циклопентанон-производным β-кетоэфиров, с которым получен соответствующий продукт с 95% выходом и 94% ee ( Enantiomeric excess, энантиомерным избытком) . С другой стороны, в случае с тетралон-производными субстратами наблюдались очень низкие (<5%) конверсии. Были проведены исследования механизма реакции, чтобы объяснить данный феномен. Предварительное образование N-SCF3 четвертичного аммония из хинина и SCF3 было первое, что предполагали авторы. Однако, не удалось найти вновь образованные соединения при контроле реакции этих двух реагентов при помощи 19F ЯМР при стандартных условиях, так что эта гипотеза была отвергнута (Схема 3).
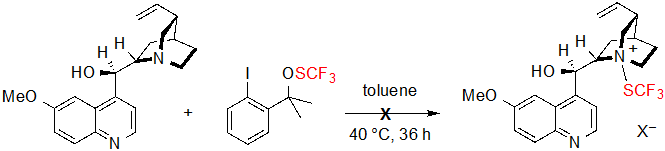
Схема 3. Ожидаемая реакция между хинином и агентом трифторметилтиолирования.
Поэтому, была предложена двойная активация как субстрата, так и агента трифторметилтиолирования посредством водорода, связывающегося с хинином (Схема 4). Такая гипотеза соответствует факту, что использование других алкалоидов хинного дерева, которые не содержат гидроксильную группу, приводит к чрезвычайно низким конверсиям. Более того, принимая во внимание, что тетралон-производные β-кетоэфиров образуют неплоские еноляты, которые наверняка не впишутся ни в данную ограниченную модель, следовательно не могут образовать двойного-активированного промежуточного соединения, данный механизм может оправдать их недостаток реакционной способности.
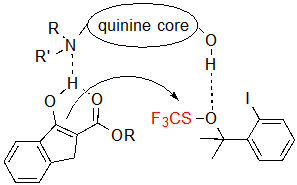
Схема 4. Двойная активация субстрата и агента трифторметилтиолирования
В результате, некоторые модификации условий реакции были необходимы, чтобы скорректировать данную проблему. Среди прочего, замена хинина на катализатор межфазного переноса на основе хинина очень помогла достичь более высоких результатов. В результате, пять SCF3 β-кетоэфиров на основе тетралона могли быть получены с хорошими выходами и приемлемыми энантиоселективностями (Схема 5). 1-Бензоциклопентанон производные, также как и субстраты на основе циклогексанона, тоже совместимы с модифицированными условиями реакции. Достаточно интересно, что указанные условия не позволили достичь удовлетворительной конверсии пяти-членного кольца β-кетоэфиров.
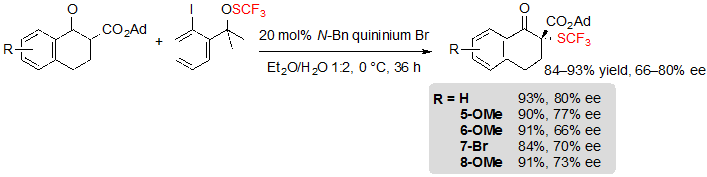
Схема 5. Ассиметричное трифторметилтиолирование адамантил β-кетоэфиров на основе тетралона
Доказано, что ациклические субстраты являются главным ограничением этих реакций, так как они показали плохую реакционную способность при использовании любого из двух предложенных катализаторов на основе хинина.
Ассиметричное трифторметилтиолирование β-кетоэфиров привлекало внимание не только группу Шена. Действительно, Rueping с коллегами сообщали о схожей методологии, основанной также применении катализа на основе алкалоидов хинного дерева [7]. В этот раз N-(трифторметилтио)фталимид использовался в качестве электрофильного источника SCF3. Используя 10% (мол.) раствор хинидина в дихлорметане при –75 °C, были получены различные (S)-инданон- производные трифторметилтиолированных β-кетоэфиров с отличными выходами и энантиоселективностью (Схема 6).
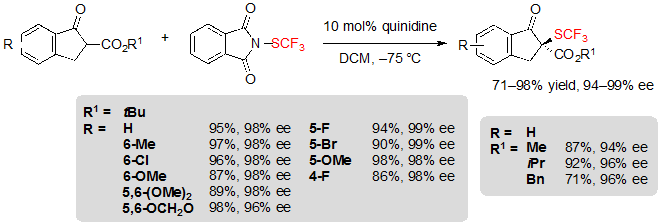
Схема 6. Ассиметричное трифторметилтиолирование β-кетоэфиров на основе инданона
Стоит подчеркнуть, что в этом случае размер сложноэфирной группы очень мало влиял и на выход, и на энантиоселективность реакции. Более того, субстрат на основе тетралона также можно преобразовать в свой SCF3 аналог, несмотря на невысокий выход (46% выход, 95% ее), увеличив температуру до 0 °C и удлинив время реакции до семи дней.
Циклопентонен-производные терт-бутил β-кетоэфиров также совместимы с условиями реакции, давая соответствующие продукты с хорошими выходами и высокой энантиоселективностью (Схема 7).
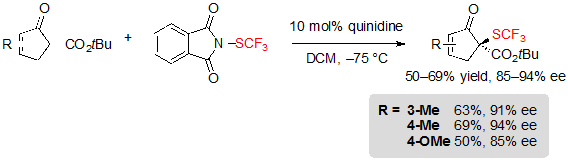
Схема 7 Ассиметричное трифторметилтиолирование β-кетоэфиров на основе циклопентенона
В конце концов, также возможно эффективно подойти к (R)-энантиомеру переключившись с хинидина на хинин псевдоэнантиомера (Схема 8).
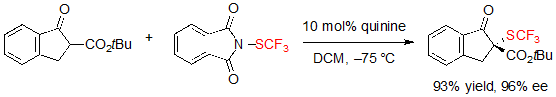
Схема 8. Подход к (R)-SCF3 β-кетоэфирам на основе инданона
В дополнении к данным органо-каталитическим подходам Gade с сотрудниками описали метод, основанный на метал-катализе для проведения реакции такого же типа. Соединение комплекса Cu-BOXMI (bis(oxazolinylmethylidene)isoindolines) и реагента трифторметилсульфената позволило получить циклические трифторметилтиолированные β-кетоэфиры с отличными показателями выходов и высокой энантиселективностью (Схема 9) [8]. Реакция совместима с инданонон-, тетралон-, циклопентенон а также с циклогексенон-производными β-кетоэфиров, и разнообразные функциональные группы такие, как метокси, метил или галоген хорошо совместимы. И снова, громозкость группы сложных эфиров мало повлияла на энантиоселективность, за исключением случая проведения реакции на производных тетралона.
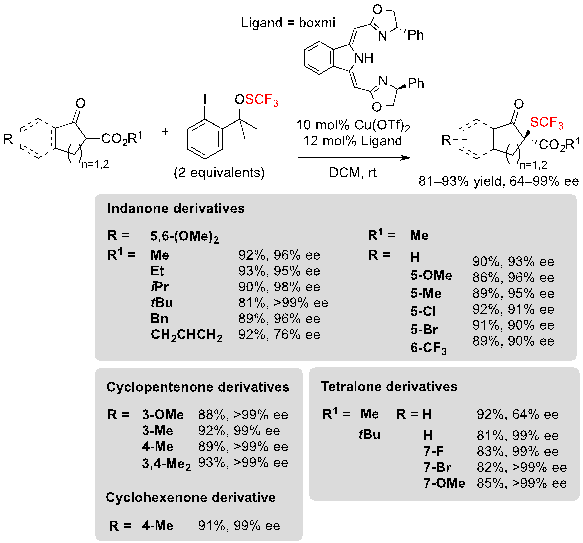
Схема 9. Катализированное медью трифторметилтиолирование различных β-кетоэфиров
Что касается механизма реакции, органокатализ только за счет действия лиганда был быстро исключен, так как ни один продукт не образовался в отсутствие солей меди. Более того, возможная прямая реакция между реагентом SCF3 и комплексом меди не была зафиксирована ни ЭПР спектроскопией, ни 19F ЯМР спектроскопией. Принимая во внимание информацию экспериментов, авторы предложили путь, отраженный в Схеме 10. Во-первых, оба карбонильных атома кислорода β-кетоэфира координируются с комплексом Cu-BOXMI. Полученное промежуточное соединение А затем депротонируется трифлатным анионом, давая сложный эфир енолята B, в который можно ввести группу SCF3 из агента трифторметилтиолирования через активацию трифторметансульфоновой кислоты. Целевой продукт получается из промежуточного соединения С после конечной стадии декоординации. Энантиоселективность процесса предположительно возникает из того факта, что Si-face сторона енолята B экранирована фенилом лиганда BOXMI и что, следовательно, электрофильный SCF3 предпочтительно подходит с положения Re-face.
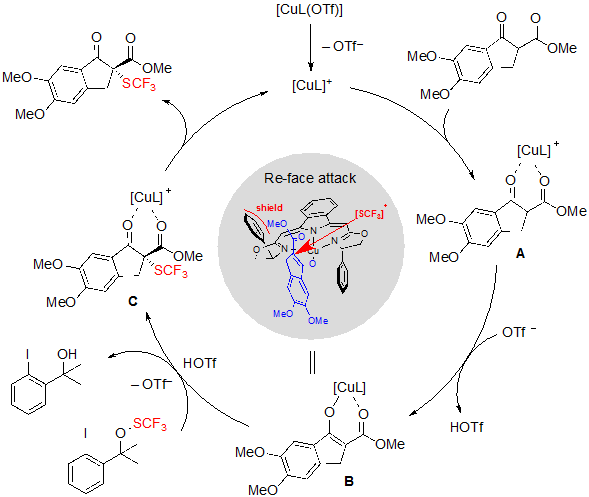
Схема 10. Механизм каталитического трифторметилтиолирования циклических β-кетоэфиров с помощью меди
Du с коллегами были также заинтересованы в ассиметричном введении SCF3 фрагмента на 1,3-дикарбонилы. В 2017 они предложили энантиоселективное трифторметилтиолирование циклоалкенона благодаря N-трифторметилтиосукцинимиду и катализатору squaramide ( амиды квадратной кислоты, 1,2-дигидроциклобутендиона), полученного из гидрохинина и D-глюкопиранозы (Схема 11) [9].
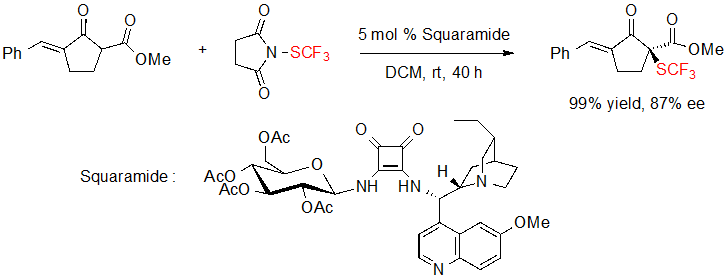
Схема 11. Энантиоселективное трифторметилтиолирование циклоалкенона катализатором squaramide.
Такие субстраты также могут реагировать с 2-меркаптобензальдегидными производными в однореакторной (реакции, которую проводят в одном реакционном сосуде без выделения промежуточных соединений) процедуре для образования SCF3 спиро (полициклическое соединение, кольца которого имеют один общий атом) циклопентанон-тиохроманов через реакцию каскада sulfur-Michael/aldol (Схема 12).
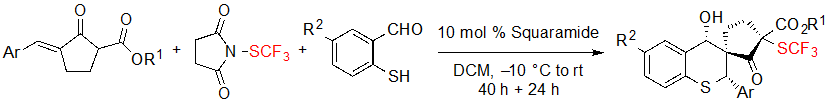
Схема 12. Однореакторный синтез SCF3 спиро циклопентанон-тиохроманов
Интересно, что производное циклоксенона было эффективно преобразовано в свой SCF3 аналог при слегка измененных условиях, но не могло участвовать в каскадной реакции (Схема 13).
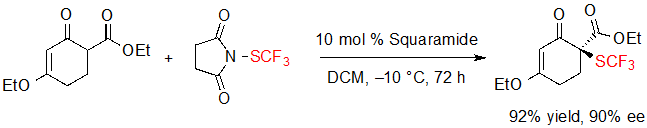
Схема 13. Энантиоселективное трифторметилтиолирование производного циклоксенона
Трифторметилтиолирование оксиндолов
Оксиндолы также являлись привилегированными субстратами для энантиоселективного введения фрагмента SCF3. Rueping и коллеги впервые сообщили о процедуре, позволяющей проводить ассиметричное трифторметилтиолирование оксиндолов, используя те же условия реакции как и те, что они предложили для β-кетоэфиров (Схема 14) [10]. Однако, хинидин должен был быть заменен на катализатор из алкалоида хинного дерева (bis-cinchona alkaloid), (DHQD)2Pyr, чтобы получить высокие показатели энантиоселективности. Различные 3-арил BOC(tert-butyloxycarbonyl)-защищенные оксиндолы, замещенные различными фрагментами на ариле, а также на конденсированном кольце гетероцикла, получали независимо от ассоциированных продуктов с выходами и энантиоселективностью от хороших до превосходных.
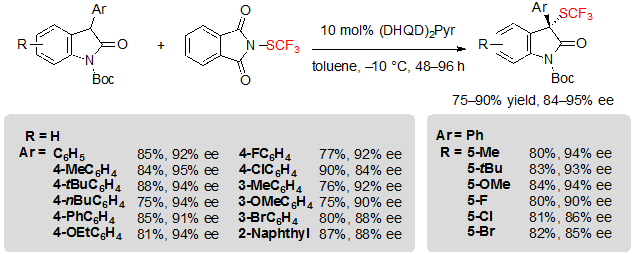
Схема 14. Ассиметричное трифторметилтиолирование 3-арил оксиндолов.
Вскоре после этого Tan, Liu и их коллеги описали похожую методику, основанную генерации электрофильного реагента трифторметилтиолирования in situ из легко доступного AgSCF3 и трихлоризоциануровой кислоты (TCCA) (Схема 15) [11]. Реакционные условия совместимы с 3-арил оксиндолами с различными вариантами замещения и предлагали ожидаемые продукты с выходами от средних до отличных и высокой энантиоселективностью после однореакторной стадии удаления защитной группы.
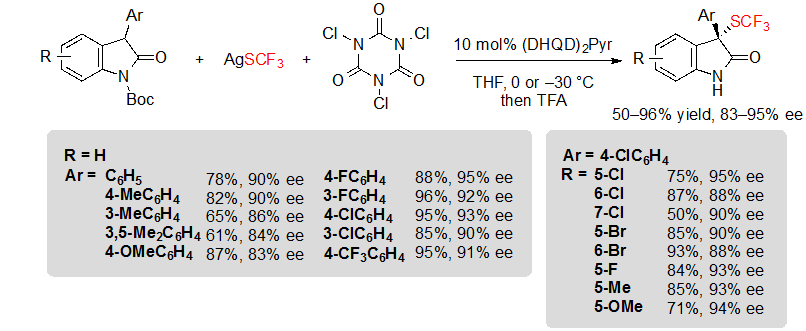
Схема 15. Ассиметричное трифторметилтиолирование 3-арил оксиндолов с генерацией in situ электрофильного SCF3 донора
Эксперименты, проводимые для идентификации реакционноспособного электрофильного SCF3 показали, что бис(трифторметил)дисульфид образовывался на месте и принимал участие в конверсии субстрата. Однако, образование трифторметилтиолированного продукта также происходило со схожей энантиоселективностью без упомянутого дисульфида, несмотря на присутствие нерастворимых частиц, получаемых из реакции TCCA и AgSCF3. К сожалению, данные вещества нельзя было идентифицировать (Схема 16).
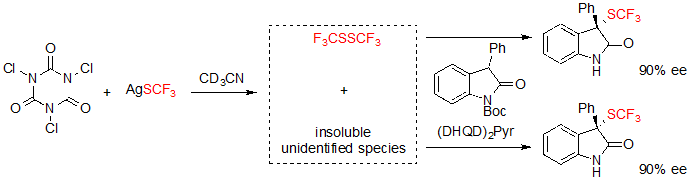
Схема 16. Исследование активных соединений реакции.
Shen с коллегами в конце концов предложили хотя и основанную на схожей концепции, но альтернативную методологию, с достойным внимания ранее неизвестным преимуществом предоставления пути к трифторметилтиолированным 3-алкил оксиндолам с отличными показателями выходов и высокой энантиоселективностью (Схема 17) [12]. Для достижения таких результатов было необходимо использовать 1.2 эквивалента трифторметилсульфената реагента Shen совместно с 20 % (мол.) хинина в диэтиловом эфире при 35 °C. Оказалось, что природа реагента трифторметилтиолирования по-настоящему влияет на реакцию; в самом деле, его замена на N-(трифторметилтио)фталимид спровоцировало значительное снижение энантиселективности. К сожалению, реакция протекала менее эффективно с 3-арилоксиндолами, предлагая сопутствующие продукты с чуть более низкой энантиоселективностью. Что касается плюсов, то было обнаружено, что другие защитные группы помимо BOC, такие как CO2Ad могут быть применены без особого влияния на величины ее.
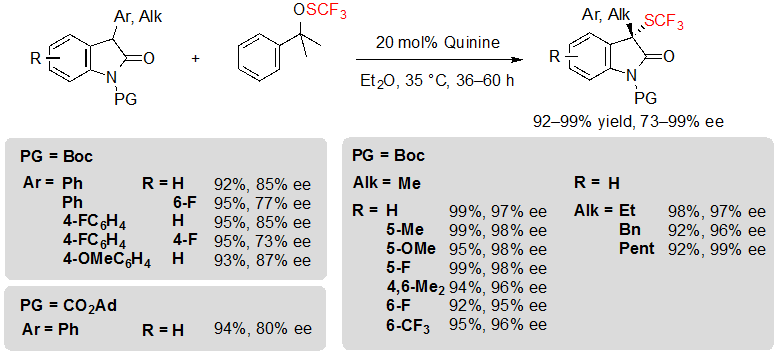
Схема 17. Ассиметричное трифторметилтиолирование 3-арил и 3-алкил оксиндолов
Реакции трифторметилтиолирования в направлении дитиокеталей
Группа под руководством Zhou заинтересовалась синтезом фрагментов дитиокеталей, так как они обещают большой потенциал в биофармацевтической химии. В данном контексте они описали ассиметричное введение SCF3 группы в 1-инданон-производные тиоэфиров используя 1.2 эквивалента N-(трифторметилтио)сукцинимида и 10 %( мол.) дигидрохинина в дихлорметане при –20 °C. После 6 дней соответствующие SCF3 продукты были выделены с превосходными показателями энантиоселективности, несмотря на скромные показатели выходов (Схема 18) [13] .
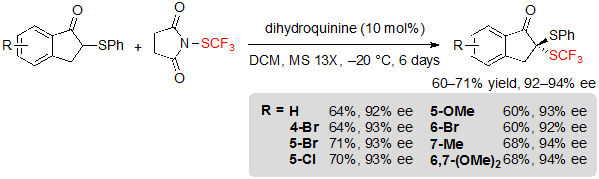
Схема 18. Ассиметричное трифторметилтиолирование 1-инданон-производных тиоэфиров
Интересно, что также явилось возможным подойти к более разнообразным дитиокеталям, полученным из оксиндола, 2-инданона или 3-бензофуранона с похожими скромными показателями выходов и хорошими ее величинами (Схема 19).
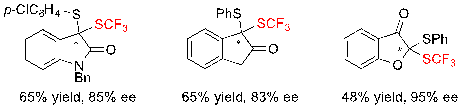
Схема 19. Подход к структурно разнообразным дитикеталям
Трифторметилтиолирование альдегидов
Во время исследования, фокусированного на трифторметилтиолировании альдегидов, Wu, Sun и коллеги попытались расширить свою методологию, развивая ее ассиметричную версию (Схема 20) [14]. К сожалению, при проведении реакции в присутствии катализатора Hayashi-Jørgensen показатель ее составлял только 11%. До настоящего момента не сообщалось о дальнейших исследованиях данной реакции.
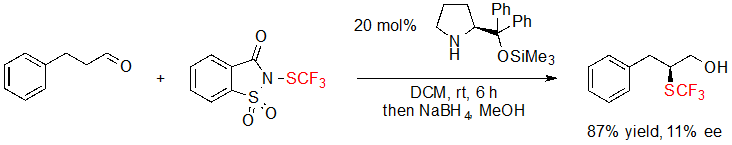
Схема 20. Ассимитричное α- трифторметилтиолирование альдегида
Каскад трифторметилтиолирование /циклизация из функционализированных алкенов
Недавно Zhao с коллегами изучали интересное энантиоселективноею трифторметилтиолирование алкенов с последующей стадией циклизации для синтеза хиральных лактонов. С этой целью использовался бифункциональный хиральный сульфидный или селенидный катализатор с вновь разработанным реагентом трифторметилтиолирования (PhSO2)2N–SCF3, аналогом хорошо известного фторирующего агента NFSI. Таким образом, используя 1.5 эквивалента указанного реагента, 0,5 эквивалентов трифлатной (трифторметансульфокислота) кислоты и 20% (моль) хирального сульфида на основе индана в дихлорметане при 0 °C, различные 4-арил замещенные (E)-but-3-enoic кислоты можно преобразовать в соответствующие лактоны через каскадную реакцию трифторметилтиолирования/лактонизации. (Схема 21) [15,16]. Небольшое снижение энантиоселективности наблюдалось у субстратов с арилами, несущими электронодонорную группу в пара положении. Таким же образом, скрытые алкены с 2-замещенным арил фрагментом привели к меньшим ее величинам. С другой стороны, когда электроноакцепторные группы были введены в пара и мета положения арила, это повляло на выход продукта, и он снизился до 55%. Арильный заместитель можно сменить на бензильную группу без особых последствий; однако, его замена на длинные алкильные цепи приводит к смеси диастереомеров и скромным показателям энантиоселективности.
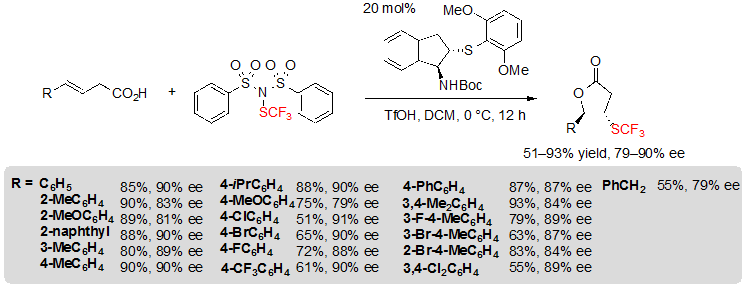
Схема 21. Лактонизация алкенов при ассиметричном трифторметилтиолировании.
Что касается механизма реакции, авторы предложили путь, представленный на Схеме 22. BOC-защищенный катализатор сначала реагирует с трифторметансульфокислотой, давая соответствующий трифлат А. SCF3 катион затем связывается с катализатором, чтобы образовать промежуточный сульфенелиум ион В через активацию трифторметансульфокислоты. В присутствие олефиновой карбоновой кислоты промежуточное вещество В легко превращается в трифторметилтиираниум (trifluoromethylthiiranium) ион С, который в конце концов атакуется кислородом карбоксильной группы, чтобы дать на выходе ожидаемый продукт и регенерировать катализатор в процессе. Данный механизм проливает свет на удобство в использовании бифункционального катализатора, который способен не только связывать SCF3 катион через атом серы, но также освободжать карбоксильную группу для стадии циклизации. Интересно, что схожие по структуре катализаторы, несущие селен вместо атома серы также могут использоваться, но при этом достигаются более низкие показатели энантиоселективности. Это можно объяснить меньшим размером атома серы, который приводит к улучшенному стерическому несоответствию вокруг SCF3катиона и соответственно к лучшему стереоконтролю.
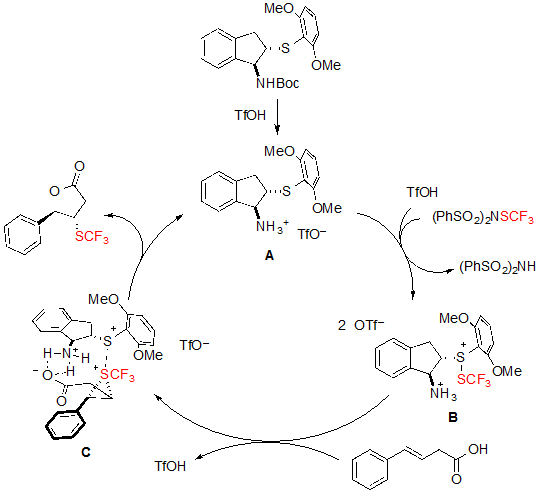
Схема 22. Механизм ассиметричной трифторметилтиоляционной лактонизации алкенов
В контексте их работы над трифторметилтиоляционной циклизацией алкенов Zhao с сотрудниками также описали подход к насыщенным SCF3 азагетероциклам, основанным на схожей концепции (Схема 23) [17].[ Для этой цели был выбран такой же реагент трифторметилтиолирования (PhSO2)2N–SCF3 и использован хиральный селенидный катализатор, несущий трифлиловую (triflyl) группу на основе индана. Как уже однажды случалось в предыдущей работе Zhao стерическое несоответствие и электронные эффекты катализатора являлись ключевыми параметрами тонкой настройки для получения высоких величин ее.
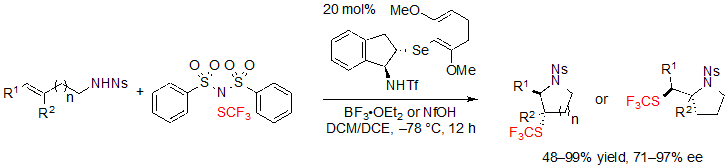
Схема 23. Ассиметрическая трифторметилтиоляционная циклизация (E)-алкенов по направлению к насыщенным SCF3 азагетероциклам
Таким образом, используя 1.5 эквивалента (PhSO2)2NSCF3, 20 % (моль) селенидного катализатора и 1.0 эквивалента BF3·OEt2 в смеми 1:1 дихлорметана и дихлорэтана при –78 °C, различные (E)-алк-3-ен-1-сульфонамиды давали ассоциированные продукты с высокими выходами, величинами dr и ee (Схема 24). Алкил- и арил-замещенные алкены также хорошо применимы. Арилалкены с электроноакцепторными группами, такими как галогены, однако, менее легко преобразовывались и было необходимо поднять температуру реакции для получения высоких выходов. С другой стороны (Z)-изомеры вступали в реакцию.
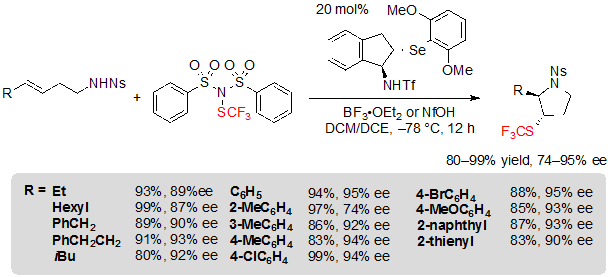
Схема 24. Ассиметрическая трифторметилтиоляционная циклизация (E)-алкенов по направлению к насыщенным пятичленным SCF3 азагетероциклам
Доказано, что Алк-4-ено-1-сульфонамиды – это субстраты, представляющие большую сложность в работе; на деле наблюдалась конкуренция между endo и exo циклизацией, а природа получаемого продукта более независима при замещении начального материала. exo продукт был в основном получен в случае конечных, алкил и тризамещенных алкенов с показателями выходов и энантиоселективностями от средних до отличных (Схема 25).
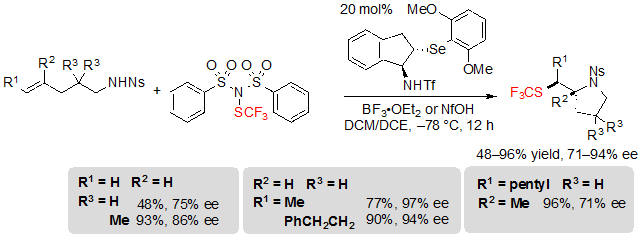
Схема 25. Ассиметрическая трифторметилтиоляционная exo циклизация (E)-алк-4-енов по направлению к пирролидинам
Образование exo продукта происходит как главный путь при проведении реакции с заместителем фенила на алкене (Схема 26).
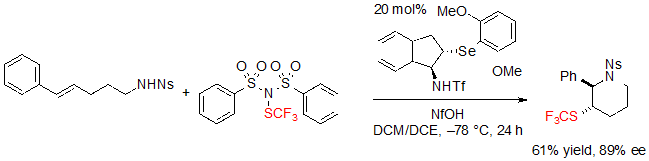
Схема 26. Ассиметрическая трифторметилтиоляционная endo циклизация (E)-алк-4-ена по направлению к насыщенному шестичленному SCF3 азагетероциклу
2.3. Хиральные реагенты трифторметилтиолирования
В 2017, Shen с коллегами предложили использование оптически чистых реагентов трифторметилтиолирования для энантиоселективного трифторметилтиолирования [18]. Производные (1S)-(–)-N-Трифторметилтио-2,10-камфорсултама ((1S)-(–)-N-Trifluoromethylthio-2,10-camphorsultam) оказались лучшими кандидатами для проведения такого типа реакций. Производным N-трифторметилтиоксазолидина тоже дана была оценка, но они дали более низкие энантиоселективности (Схема 27).
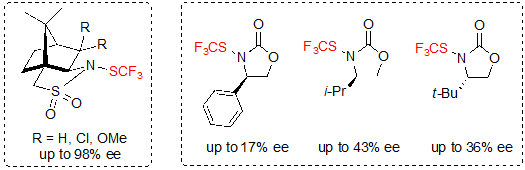
Схема 27. Хиральные реагенты трифторметилтиолирования (ee величины для реакции с β-кетоэфирами).
Данные реагенты также совместимы с различными субстратами. Например, в присутствие 0.1 эквивалента K2CO3 в тетрагидрофуране при –40 °C, пяти и шести-членные кольцевые β-кетоэфиры позволили получить соответствующие SCF3 продукты с показателями выходов от хороших до отличных с высокой энантиоселективностью, до 98% (Схема 28). Несколько функциональных групп (R) и различные группы сложных эфиров (R1) также хорошо переносятся. Однако, было необходимо увеличить температуру для достижения удовлетворительной конверсии при проведении реакции на субстратах, несущих электроно-акцепторные R группы.
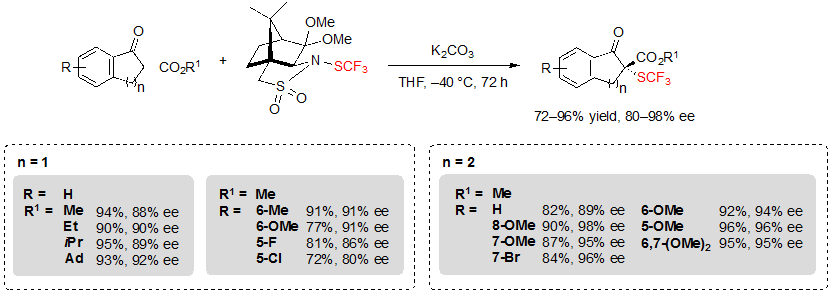
Схема 28. Ассиметричное трифторметилтиолирование циклических β-кетоэфиров на основе инданона и тетралона
Таким же образом, разнообразные 3-арилоксиндолы с готовностью преобразовывались в свои трифторметилтиолированные аналоги после небольшой модификации условий реакции. Примечательно, что K2CO3 был заменен на Cs2CO3 а тетрагидрофан – на диэтиловый эфир (Схема 29). В общем-то, условия реакции снова оказались эффективными, несмотря на функциональные группы, переносимые арильной группой, или характер защитной группы, соединенной с азотом. Однако, в уникальном случае с оксиндолом, несущим цианометил заместитель в пара положении своего 3-арильного кольца, наблюдалось падение энантиоселективности. Несмотря на это данный вопрос можно решить, заменив трифторметилтиолирующий реагент на другой аналог камфоросультама. Указанный реагент также позволял достичь более высоких показателей ее для субстратов, прошедших оценку, и даже провести реакцию на 3-метил оксиндоле со средней энантиоселективностью.
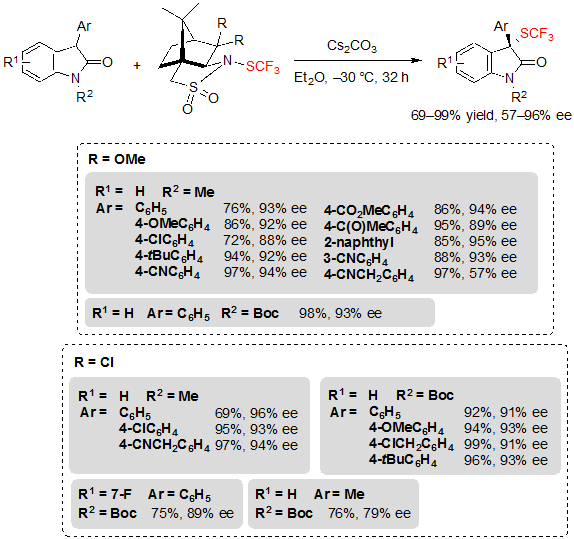
Схема 29. Ассиметричное трифторметилтиолирование 3-арилоксиндолов
В конечном итоге также было возможным подступиться к структурно новым хиральным 2-SCF3-2-арил бензофуран-2(3H)-ам с показателями выходов от скромных до высоких и величинами ее в диапазоне 64–92% (Схема 30).
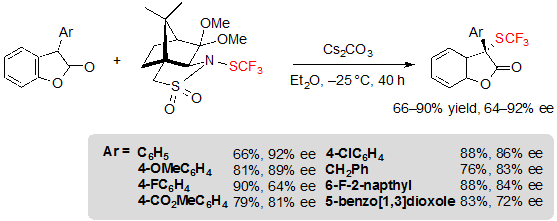
Схема 30. Ассиметричное трифторметилтиолирование 2-арил бензофуран-2(3H)-ов
3. Блочный принцип конструирования
По сравнению с методами прямой энантиоселективной трифторметилтиоляции блочный принцип конструирования хиральных центров, несущих группу SCF3, совсем не распространен. В настоящее время можно обнаружить всего лишь два недавних сообщения о стереоконтролируемом хиральном образовании SCF3 соединений.
Первый подход, внедренный Wang с коллегами – это вариация реакции Doyle-Kirmse [19]. Она проходит стадию энантиоселективной [2,3]-сигматропной перегруппировки илида (ylide) сульфония, который генерируется из аллил или пропаргил трифторметил сульфидов и карбенов металлов, полученных из реакции диазоэфира и катализатора металла, несущего хиральные лиганды. Данная методология впервые была разработана с использованием катализатора родия (II), Rh2(S-DOSP)4, в n-пентане, при –30 °C. Широкий диапазон ароматических диазоэфиров применяли в этой реакции, давая соответствующие хиральные (R)-трифторметил сульфиды с показателями выходов от хороших до отличных и высокими величинами ее (Схема 31). Интересно, что громозкость группы сложного эфира серьезно влияла на энантиоселективность реакции; на самом деле, величины ее упали, при увеличении стерический объема группы сложного эфира. Электронные эффекты также оказались важны; диазоэфиры, несущие арилы с электронодонорными группами были преобразованы со сниженными выходами и энантиоселективностью. Данный феномен можно отнести к меньшей стабильности таких веществ, которые в связи с этим более склонны к тому, чтобы быть поглощенными побочными реакциями такими, как димеризация.
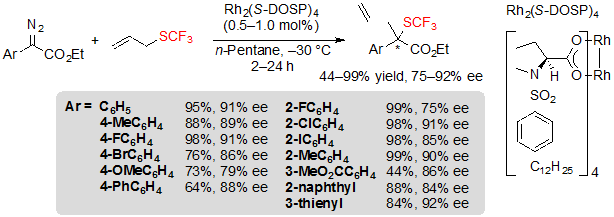
Схема 31. Rh-катализованная [2,3] сигматропная перегруппировка из ариловых диазоэфиров и аллилового трифторметилсульфида
Реакцию также можно провести эффективно на винил-диазоацетатах и более высокие ее величины были достигнуты на самом деле с данными субстратами (Схема 32).
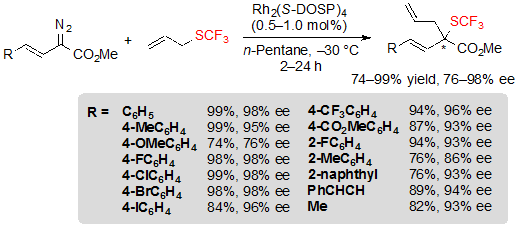
Схема 32. Rh-катализованная [2,3] сигматропная перегруппировка из виниловых диазоэфиров и аллилового трифторметилсульфида
В конце концов, стало также возможным заменить аллиловый трифторметил сульфид на пропагил трифторметил сульфид и прореагировать его со схожими арил- и винил-диазоэфирами для того, чтобы иметь доступ к соответствующим аллениловым сульфидам. Реакция проходила с показателями выходов и энантиоселективности от скромных до хороших. (Схема 33).
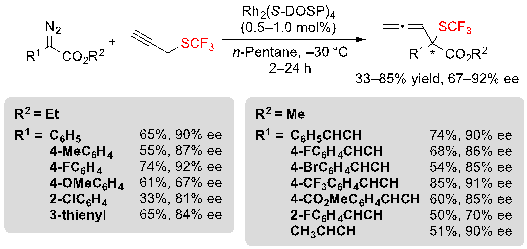
Схема 33. Rh-катализованная [2,3] сигматропная перегруппировка из диазоэфиров и пропагил трифторметилсульфида
Медные(I) катализаторы имеют преимущество перед комплексами родия в том, что они более дешевы. При использовании Cu(CH3CN)4PF6, соединенногого с хиральным лигандом, реакция продемонстрировала хорошую эффективность и позволила получить ожидаемые продукты с существенно идентичной энантиоселективностью (Схема 34). Реакция имеет те же ограничения, что и с родиевым катализом: диазоэфиры, несущие богатые электронами ароматические кольца, давали низкие выходы и относительно высокие показатели энантиоселективности. С другой стороны, в данных условиях винил-диазоацетаты давали низкие величины ее, а конверсия пропагил сульфидов шла особенно медленно.
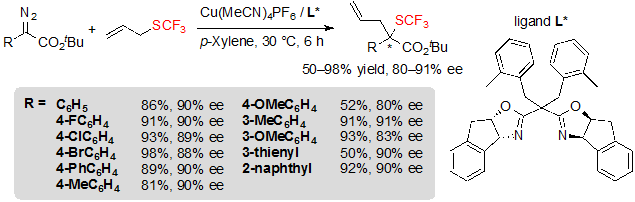
Схема 34. Cu-катализованная [2,3] сигматропная перегруппировка из диазоэфиров и аллилового трифторметилсульфида
Как упоминалось ранее в тексте, рассматриваемая реакция является ассиметричным примером реакции Doyle-Kirmse. Первый шаг упомянутой реакции состоит из образования хирального металлического карбена с последующим его координироанием с атомом серы сульфида аллила. Полученный ylide сульфония затем подвергается синхронной [2,3]-сигматропной перегруппировке, переносящей хиральность с серы на углерод (Схема 35). Образование свободного ylide – это главный шаг в отношении энантиоселективности. Гипотеза, что реакция может протекать через Rh(II)-ассоциированный ylide была закреплена несколькими контрольными экспериментами.
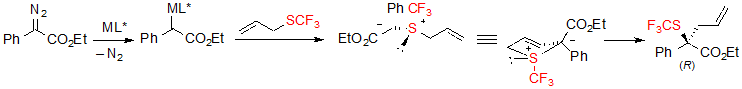
Схема 35. Механизм реакции
Zhao с коллегами фокусировались на другом типе блочного конструирования и описали ассиметрические органофосфин-катализованные реакции Манниха (Mannich-type) на N-BOC иминах, предоставив доступ к тетразамещенным карбоновым стереоцентрам, несущим циано и группу SCF3 [20]. В данном исследовании арил 2-(трифторметилтио)ацетонитрилы были выбраны в качестве субстратов. Лучшие результаты получены при использовании 1.0 эквивалента SCF3 блока, 2.0 эквивалентов BOC-защищенных иминов, 10 % (моль) хирального фосфина и 10% (моль) метил акрилата. Ожидаемые продукты были получены со скромными dr (до 78:22) и величинами ее от скромных до высоких, соответственно выходы, относящиеся к обоим диастереомерам ограничены, до 68% для основного диастереомера (Схема 36). Те же выходы были получены независимо от схемы поведения замены субстратов. Что касается энантиоселектиновности, электронодонорные группы, несущие Ar группу имина поспособствовали ощутимо увеличению величин ее, в то время как электроно-акцепторные группы такие, как галогены дали более худшие результаты. Также оказалось, что иминозащитная группа тоже достаточно влиятельна. На самом деле, реакция не протекала при использовании p-метоксифенила, p-толуолсульфонила, 1,1-дифенилметенила или дифенилфосфонила, хотя она прекрасно сработала с N-CBz имином, хоть и с более низкой величиной ее.
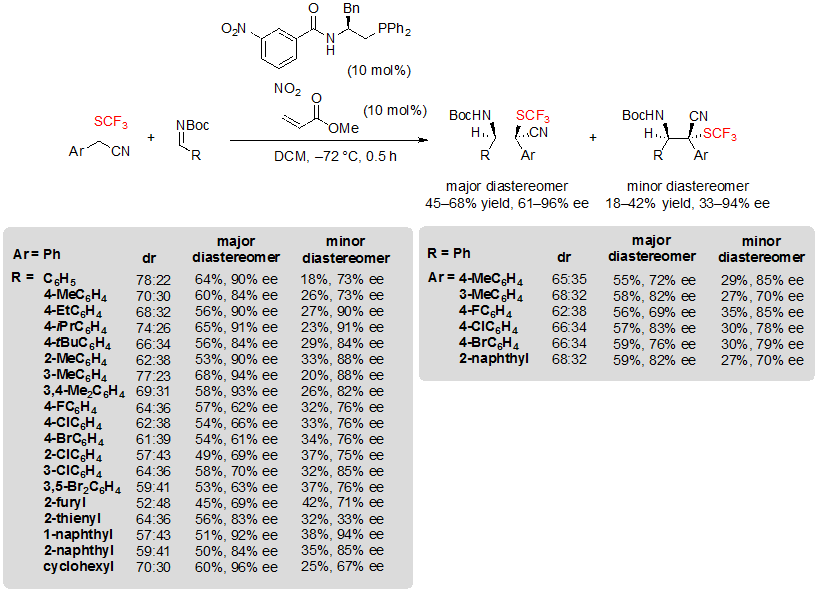
Схема 36. Образование CF3S-содержащих тетразамещенных карбоновых стереоцентров через энантиоселективные реакции Манниха.
Что касается механизма реакции, авторы предложзили путь, представленный на Схеме 37. Сначала два компонента каталитической системы – хиральный фосфин и метил акрилат образуют цвиттер-ионный аддукт А, как и подтверждено ESI-MS, которая выступает в качестве мягкой хиральной основы BrØnsted. Этот енолят фосфония А депронирует SCF3-субстрат, чтобы образовать ион-пару B, которая затем может атаковать BOC-имин, образуя промежуточное вещество D через переходное состояние С. Затем происходит обмен протонами между SCF3структурным блоком и промежуточным веществом D, образуя смесь диастереомеров с регенерирацией вещества B. Данный механизм подчеркивает важность нитрильной группы на SCF3 субстрате. На самом деле, нитрил необходим для эффективного протекания реакции, потому что сильно способствует депротонированию субстрата. Также стоит упомянуть, что реакция не может протекать при замене циано группы на молекулы сложных эфиров.
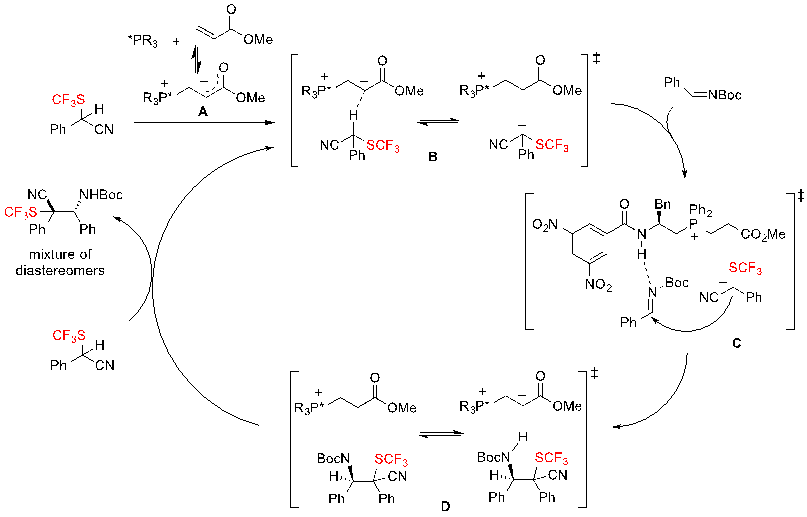
Схема 37. Предполагаемый механизм энантиоселективной реакции Манниха
4. Перспективы
Ассиметрический синтез трифторметиолированных молекул – это зарождающаяся область исследования органической химии фтора. С 2013 принципы прямого внедрения электрофильного SCF3 фрагмента через органокатализ, метал-катализованные реакции или с помощью хиральных реагентов описаны в публикациях, оказавших большое влияние. Недавно, было сообщено об использование структурных блоков, содержащих фрагмент SCF3, для стереоконтролируемого получения хиральных SCF3 соединений. На этой стадии все еще необходимо продолжение исследований по двум направлениям:
(i) поиск альтернативных более эффективных, дешевых и более простых в работе реагентов, которые могут выступать как донор SCF3 группы.
(ii) развитие новых ассиметричных синтезов с помощью инновационного подхода.
Эти новые инструменты позволят достичь новых молекулярных архитектур с SCF3 и даже более высоких стереоселективностей. Мы уверены, что текущий уровень развития, детализированный в этом обзоре, должен стимулировать дальнейшие разработки в области хиральных трифторметилтиолированных молекул для медицины, науки о материалах и других областей нашей повседневной жизни.
Благодарности
Наше исследование было поддержано Французским Центром Научных Исследований (Centre National de la Recherche Scientifique (CNRS)), Университетом г. Руана (University of Rouen), институту INSA Rouen, Labex SynOrg (ANR-11-LABX-0029). Hélène Chachignon благодарит Министерство высшего образования и научных исследований Франции за докторскую стипендию. Evgeniy V. Kondrashov благодарен Посольству Франции в Москве за Исследовательский Грант Мечникова.
Литература
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa, H. Liu, Chem. Rev. 2016, 116, 422-518.
- a) V. Bizet, T. Besset, J.-A. Ma, D. Cahard, Curr. Top. Med. Chem. 2014, 14, 901-940; b) J.-A. Ma, D. Cahard, Chem. Rev. 2008, 108, PR1-PR43; c) J. Nie, H.-C. Guo, D. Cahard, J.-A. Ma, Chem. Rev. 2011, 111, 455-529.
- a) F. Toulgoat, T. Billard, in Modern Synthesis Processes and Reactivity of Fluorinated Compounds (Eds.: F. R. Leroux, A. Tressaud), Elsevier, 2017, pp. 141-179; b) X. H. Xu, K. Matsuzaki, N. Shibata, Chem. Rev. 2015, 115, 731-764.
- H. Chachignon, D. Cahard, Chinese J. Chem. 2016, 34, 445-454.
- X. Shao, X. Wang, T. Yang, L. Lu, Q. Shen, Angew. Chem., Int. Ed. 2013, 52, 3457-3460.
- X. Wang, T. Yang, X. Cheng, Q. Shen, Angew. Chem., Int. Ed. 2013, 52, 12860-12864.
- T. Bootwicha, X. Liu, R. Pluta, I. Atodiresei, M. Rueping, Angew. Chem., Int. Ed. 2013, 52, 12856-12859.
- Q.-H. Deng, C. Rettenmeier, H. Wadepohl, L. H. Gade, Chem. Eur. J. 2014, 20, 93-97.
- B.-L. Zhao, D.-M. Du, Org. Lett. 2017, 19, 1036-1039.
- M. Rueping, X. Liu, T. Bootwicha, R. Pluta, C. Merkens, Chem. Commun. 2014, 50, 2508-2511.
- X. L. Zhu, J. H. Xu, D. J. Cheng, L. J. Zhao, X. Y. Liu, B. Tan, Org. Lett. 2014, 16, 2192-2195.
- T. Yang, Q. Shen, L. Lu, Chinese J. Chem. 2014, 32, 678-680.
- K. Liao, F. Zhou, J. S. Yu, W. M. Gao, J. Zhou, Chem. Commun. 2015, 51, 16255-16258.
- L. Hu, M. Wu, H. Wan, J. Wang, G. Wang, H. Guo, S. Sun, New J. Chem. 2016, 40, 6550-6553.
- X. Liu, R. An, X. Zhang, J. Luo, X. Zhao, Angew. Chem., Int. Ed. 2016, 55, 5846-5850.
- J. Luo, X. Liu, X. Zhao, Synlett 2017, 28, 397-401.
- J. Luo, Y. Liu, X. Zhao, Org. Lett. 2017, 19, 3434-3437.
- H. Zhang, X. Leng, X. Wan, Q. Shen, Org. Chem. Front. 2017, 4, 1051-1057.
- Z. Zhang, Z. Sheng, W. Yu, G. Wu, R. Zhang, W.-D. Chu, Y. Zhang, J. Wang, Nat Chem 2017, DOI: 10.1038/nchem.2789.
- G. Zhao, L. Xu, C. Zheng, H. Wang, Adv. Synth. Catal. 2017, DOI: 10.1002/adsc.201700321.
Статья рекомендована к публикации членом редколлегии д.х.н. С.М. Игумновым
Fluorine Notes, 2017, 113, 5-6


