Поступило в редакцию: сентябрь 2015
УДК 547.057, 547.7/.8, 547.1'1
Fluorine Notes, 2015, 102, 3-4
Метод введения фторсодержащих заместителей в структуру порфирина путем нуклеофильного замещения атома фтора в мезо-тетракис(пентафторфенил)порфирине и пентафторбензальдегиде на полифторалифатические спирты
аЕ.В. Беляева*, аА.Л. Сиган, бЯ.Э. Дружинина, аН.С. Иконников, аН. Д. Чкаников
aФедеральное государственное бюджетное учреждение науки Институт элементоорганических
соединений им. А.Н. Несмеянова Российской академии наук, 119991, ГСП-1, Москва, В-334, ул.
Вавилова, д. 28
e-mail: faftor.belyaeva@mail.ru
bРоссийский химико-технологический университет им. Д.И.Менделеева, 125047, Российская
Федерация, 125047, г. Москва, Миусская пл., 9,
Факс: +7-495 135-5085
Аннотация: Рассмотрены варианты получения мезо-тетракис(тетрафторфенил)порфиринов, дополнительно модифицированных полифторалкоксильными заместителями. Один из вариантов сборки макроцикла подразумевает синтез и конденсацию ряда полифторалкоксизамещенных бензальдегидов, полученных путем замещения атомов фтора в пара- и орто-положениях в исходном пентафторбензальдегиде на полифторалифатические спирты. В качестве альтернативного варианта осуществлено прямое замещение атомов фтора в мезо-тетракис(пентафторфенил)порфирине на остатки полифторалифатических спиртов.
Ключевые слова: фторсодержащие порфирины, мезо-тетракис(пентафторфенил)порфирин, полифторалифатические спирты, пентафторбензальдегид, нуклеофильное замещение атома фтора.
Соединения порфиринового ряда играют ключевую роль во многих химических и биохимических процессах, а возможность введения в порфириновый цикл заместителей различной природы существенно расширяет рамки их потенциального применения в различных областях химической технологии в качестве катализаторов, компонентов фотоактивных материалов и лекарственных средств. В последнее время значительное внимание уделяется изучению фторсодержащих порфиринов в связи с развитием технологии FBS (Fluorous Biphasic System) [1,2], работами над созданием фотоэлектроактивных материалов, дополнительно обладающих гидрофобными характеристиками [3,4] и расширением круга эффективных фотосенсибилизаторов, потенциально применимых в составе препаратов для фотодинамической терапии рака (ФДТ) [5]. Исследования в этих направлениях показали, что растворимость в перфторуглеродных средах и гидрофобность обеспечиваются введением в молекулу порфирина заместителей (не менее 4-х) с длиной перфторалкильной цепи 3-8 атомов углерода [1,2], в то время как на редокс-свойства заметно влияет наличие уже трифторметильных заместителей [6].
На данный момент основными способами получения фторированных порфиринов являются каталитическое алкилирование перфторалкилиодидами или введение фторсодержащих фрагментов путем нуклеофильного замещения [7]. При этом в связи с расширением области применения фторированных порфиринов поиск новых методов их синтеза не утратил своей актуальности. Наш подход к решению этой задачи заключается в использовании полифторалкильных спиртов в реакциях замещения атома/атомов фтора под действием ряда оснований в пентафторбензальдегиде с последующей конденсацией полученных соединений в макроцикл (Метод А) или же аналогичное замещение активированных атомов фтора в.ароматических кольцах мезо-тетракис(пентафторфенил)порфирина (Метод Б).
Альдегид-пиррольная конденсация полифторалкоксизамещенных бензальдегидов (Метод А)
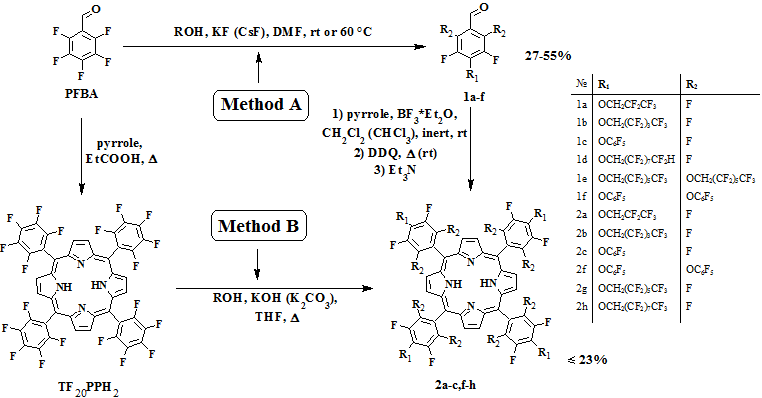
Замещение активированного атома фтора в пара-положении пентафторбензальдегида (PFBA) ранее было продемонстрировано на примере ряда фенолов [8], где для образования соответствующих фенолятов использовались такие специфические основания как фториды калия или цезия. Данная методика была использована нами в качестве базовой для введения в молекулу PFBA остатков полифторсодержащих спиртов, обладающих как и фенолы сравнительно высокой кислотностью (рКа ~12-13). Действительно, было показано, что взаимодействие PFBA с эквимолярными количествами различных полифторалифатических спиртов или пентафторфенола приводит к образованию полифторалкокси– и феноксизамещенных бензальдегидов (1a-d). Реакции проводились в диметилформамиде при комнатной температуре до полной конверсии исходного альдегида (по данным ТСХ). Следует отметить, что в случае трехкратного избытка соответствующих спиртов в этих условиях замещение атомов фтора в орто-положениях PFBA не наблюдалось независимо от времени реакции. В то же время возможность образования соответствующих 2,4,6-замещенных бензальдегидов (1e,f) с хорошими выходами была продемонстрирована при повышении температуры реакции до 60°С.
Альдегид-пиррольная конденсация полученных полифторфенокси – и алкоксизамещенных бензальдегидов (1a-c) проводилась по классическому методу Линдси [9], рекомедуемому для синтеза порфиринов из альдегидов с объемными заместителями. Реакции проводились в дихлорметане при комнатной температуре в присутствии эфирата трехфтористого бора и с использованием DDQ в качестве окислителя. Соответствующие симметричные порфирины (2a-c) были получены с хорошими для этого класса соединений выходами (10-20%) и охарактеризованы методами ЯМР- и масс-спектроскопии.
Согласно литературным данным наличие у бензальдегидов в 2-м и/или 6-м положении заместителей даже небольшого размера (например метил- или метокси-группы) значительно затрудняет возможность образования порфиринов по упомянутому выше методу. Конденсация орто-замещенных бензальдегидов с пирролом с образованием порфирина требует использования сокатализатора (этанола), что было показано на примере мезитилальдегида [10]. Использование этой методики для конденсации альдегида (1f) с пирролом привело к получению соответствующего порфирина (2f), но выход продукта не превышал 2%. Попытка провести реакцию с альдегидом (1e) оказалась безуспешной, что может быть связано с большим объемом заместителей (1,1-Н,Н-перфторгептилокси- по сравнению с пентафторфенокси-группой) или с низкой растворимостью промежуточных соединений в хлороформе, наблюдаемой нами в обоих случаях.
Замещение активированных атомов фтора в ароматических кольцах мезо-тетракис(пентафторфенил)порфирина на полифторалкоксильные фрагменты (Метод Б) [11].
Как уже отмечалось, методом введения полифторалифатических заместителей в молекулу порфирина также может служить взаимодействие мезо-тетракис(пентафторфенил)порфирина (TF20PPH2) с различными фторсодержащими N,S,O-нуклеофилами под действием оснований [7]. В то же время примеров использования в этих реакциях длинноцепочечных полифторалифатических спиртов в литературе известно не было, поэтому мы взяли за основу методику, предложенную для их нефторированных аналогов [12].
Реакции замещения проводились нагреванием TF20PPH2 в тетрагидрофуране с полифторалифатическими спиртами в присутствии гидроксида или карбоната калия (см. Схему, Метод Б). Наилучшие результаты были получены при использовании HOCH2C6F13 и HOCH2C8F17, тетразамещенные порфирины (2g,h) были выделены с выходами 35% и 47% соответственно. В качестве побочных продуктов данной реакции было отмечено образование моно-, ди- и три-замещенных порфиринов (по данным ЯМР и масс-спектроскопии), затрудняющих выделение и очистку тетразамещенного продукта. Проведение реакции со значительным избытком полифторалифатического спирта (на примере HOCH2(CF2)2H) и увеличение времени реакции не приводило к селективному получению тетра-замещенного порфирина. Однако при этом в реакционных смесях по данным масс-спектроскопии наблюдались продукты замещения атомов фтора не только в пара-, но и в орто- или мета- положениях пентафторфенильных фрагментов.
Экспериментальная часть
Спектры ЯМР 1H и 19F регистрировали на спектрометрах Bruker AMX-400 и AMX-300 с частотой 400.13 и 376.50 MГц при 20 0С, отнесение сигналов проведено относительно сигнала остаточных протонов растворителя (CDCl3) и трифторуксусной кислоты (TFA) в качестве внешнего стандарта.
Масс-спектры ESI (1) и APCI (2) регистрировали на тандемном динамическом масс-спектрометре Finnigan LCQ Advantage. Распыляющий и вспомогательный газ азот с потоками 10/0 (1) и 70/10 (2). Температура испарителя 400 °С. (2). Температура трансферного капилляра 150 оС , напряжение поля между иглой и противозлектродом 4.50 (1) и 6.0 kV (2). Образцы в растворе ацетонитрила 10-4 моль/литр вводили в ионный источник со шприцевого ввода со скоростью потока ацетонитрила 50 ( 1) и 350 (2) мкл/мин через инжектор Reodyne с петлей на 5 мкл. Масс-спектры (EI-DIP) регистрировали на масс-спектрометре Finnigan Polaris Q, энергия ионизирующего излучения 70эВ, способ ввода образца - прямой ввод.
Ход реакций контролировали методом ТСХ на пластинках Merck Kieselgel 60 F254. Для колоночной хроматографии использовали силикагель (MN Kieselgel 60) и окись алюминия (нейтральная, 100-200мкм). Элементный анализ был выполнен в лаборатории элементного анализа ИНЭОС РАН.
Реактивы и растворители: Тетрагидрофуран (ТГФ), триэтиламин (Et3N) и диметилформамид (ДМФА) осушали по стандартным методикам. Хлороформ и хлористый метилен для синтеза порфиринов осушали над CaCl2 и CaH2 соответственно и перегоняли. Пиррол и эфират трехфтористого бора перегоняли перед использованием. Фториды калия и цезия прокаливали и хранили в герметичной таре. Полифторалифатические спирты и пентафторбензальдегид использовались производства фирмы “P&M Invest”. Мезо-тетракис(пентафторфенил)порфирин получали исходя из пентафторбензальдегида и пиррола по литературной методике [13].
4-(1,1-Н,Н-Перфторпропил-1-окси)-2,3,5,6-тетрафторбензальдегид (1a)
К раствору 1,1-H,H-перфторпропан-1-ола (3.37 г, 23 ммоля) в ДМФА (40мл) прибавляли пентафторбензальдегид (2.65 г, 20 ммолей) и KF (2.65 г, 50 ммолей), перемешивали при комнатной температуре 5 часов (ход реакции контролировали по ТСХ). В реакционную массу добавляли 70мл диэтилового эфира, промывали насыщенным водным раствором NH4Cl (2 x 40мл) и водой (2 х40 мл). Органическую фазу сушили над CaCl2, после удаления растворителя получали частично кристаллизующуюся вязкую жидкость желтого цвета. После очистки продукта флеш-хроматографией (окись алюминия, гексан-хлороформ = 1:1) получали 2.77 г жидкости светло-желтого цвета (выход 38%). 1H ЯМР (CDCl3, δ, м.д.): 4.78 (т, 2H, J=12.08 Гц, OCH2), 10.20 (с, 1H, СНО). 19F ЯМР (CDCl3, δ, м.д.): -78.85 (с, 2F, CAr-Fорто), -68.04 (c, 2F, CAr-Fмета), -47.35 (с, 2F, CF2), -6.34 (c, 3F, CF3). Вычислено для C10H3F9O2 (%): C, 36.83; H, 0.93; F: 52.43. Найдено (%): C, 36.48; H: 1.02; F, 52.12.
4-(1,1-Н,Н-перфторпентил-1-окси)-2,3,5,6-тетрафторбензальдегид (1b)
К раствору 1,1-H,H-перфторпентан-1-ола (4.21 г, 17 ммолей) в ДМФА (40мл) прибавляли пентафторбензальдегид (3.00 г, 15 ммолей) и KF (2.05 г, 30 ммолей), перемешивали при комнатной температуре 5 часов (ход реакции контролировали по ТСХ). После охлаждения в реакционную массу добавили 70мл диэтилового эфира, промывали насыщенным водным раствором NH4Cl (2 x 40мл) и водой (2 х 40мл). Органическую фазу сушили над CaCl2, после удаления растворителя получали частично кристаллизующуюся вязкую жидкость желтого цвета. После очистки продукта флеш-хроматографией (окись алюминия, гексан-хлороформ = 1:1) получали 1.97 г жидкости светло-желтого цвета (выход 27%). 1H ЯМР (CDCl3, δ, м.д.): 4.82 (т, 2H, J = 2.72 Гц, OCH2), 10.23 (с, 1H, СНО). 19F ЯМР (CDCl3, δ, м.д.): -78.63 (д, 2F, J = 11.00 Гц, CAr-Fорто), -67.79 (дд, 2F, J1 = 11.00 Гц, J2 = 8.25 Гц, CAr-Fмета), -49.03 (c, 2F, CF2), -46.78 (c, 2F, CF2), -43.57 (c, 2F, CF2), -3.73 (c, 3F, CF3). Масс-спектр (EI-DIP), m/z: 477.1 [M+H]+, 459.1 [C4F9CFCHOC6F4CHF]+, 193.2 [M-C5F11CH2]+, 151.2 [CF3CFHCF2]+.
4-(1,1,9-H,H,H-нонил-1-окси)-2,3,5,6-тетрафторбензальдегид (1d)
К раствору 1,1,9-H,H,H-перфторнонан-1-ола (8.64 г, 20 ммолей) в ДМФА (20мл) прибавляли пентафторбензальдегид (3.92 г, 20 ммолей), KF (2.32 г, 40 ммолей) и перемешивали при комнатной температуре в течение 10 часов (ход реакции контролировали по ТСХ). После охлаждения в реакционную массу добавляли диэтиловый эфир (70мл), промывали насыщенным водным раствором NH4Cl (2 x 40мл) и водой (2 х 40мл). Органическую фазу сушили над CaCl2,после удаления растворителя получали частично кристаллизующуюся вязкую жидкость желтого цвета. После очистки продукта флеш-хроматографией (силикагель, элюент - этилацетат) получали 6.72 г твердого вещества бледно-желтого цвета (выход 55%). ЯМР 1Н (CDCl3, δ, м.д.): 4.82 (т, 2H, J1 = 12.72 Гц, J2 = 12.47 Гц, OCH2), 6.08 (тт, 1H, J1 = 51.84 Гц, J2 = 5.14 Гц, CF2H), 10.27 (с, 1H, CHO). ЯМР 19F (CDCl3, δ, м.д.): -78.03 (дд, 2F, J1 = 11.97 Гц, J2 = 8.98 Гц, CAr-Fорто), -67.10 (дд, 2F, J =8.98 Гц, CAr-Fмета), -59.34 (с, 2F, CF2), -51.63 (с, 2F, CF2), -45.64 (с, 2F, CF2), -45.44 (с, 2F, CF2), -44.29 (с, 6F, CF2CF2CF2), -42.92 (с, 2F, CF2). Масс-спектр (EI-DIP), m/z: 608.3 [M]+, 193.2 [M-HCF2C7F14CH2]+.
4-(пентафторфенилокси)-2,3,5,6-тетрафторбензальдегид (1c)
К раствору пентафторфенола (1.84 г, 10 ммолей) в ДМФА (10мл) прибавляли пентафторбензальдегид (1.96 г, 10 ммолей) и CsF (2.84 г, 20 ммолей) и перемешивали смесь при комнатной температуре в течение 2-х часов. Реакционную массу поместили в воду, экстрагировали этилацетатом, органическую фазу сушили над CaCl2, после удаления растворителя получали частично кристаллизующееся масло желтого цвета. После перекристаллизации полученного продукта из диэтилового эфира получено 1.81 г твердого вещества бледно-желтого цвета (выход 50%). ЯМР 1Н (CDCl3, δ, м.д.): 10.28 (с, 1H, CHO). ЯМР 19F (CDCl3, δ, м.д.): -83.26 (т, 2F, J = 20.53 Гц, C6F5O (мета-F)), -80.13 (т, 1F, J = 21.80 Гц, C6F5O (пара-F)), -78.12 (д, 2F, J = 10.90 Гц, C6F4CHO (мета-F)), -77.89 (д, 2F, J = 19.62 Гц, C6F5O (орто-F)), -66.52 (дд, 2F, J1 = 10.90 Гц, J2 = 8.72 Гц, C6F4CHO (орто-F)). Вычислено для C13HF9O2 (%): C, 43.36; H, 0.28. Найдено (%): C, 43.25; H, 0.28.
2,4,6-три(1,1-H,H-перфторгептил-1-окси)-3,5-дифторбензальдегид (1e)
К раствору 1,1-H-перфторгептан-1-ола (11.55 г, 33 ммоля) в сухом ДМФА (15мл) прибавляли пентафторбензальдегид (1.96 г, 10 ммолей) и KF (3.83 г, 66 ммолей) при интенсивном перемешивании. Реакционную смесь нагревали до 60°С и выдерживали при такой температуре 124 часа (ход реакции контролировали по ТСХ). После охлаждения в реакционную массу добавили 50 мл диэтилового эфира, промывали насыщенным водным раствором NH4Cl (2 x 30мл) и водой (2 х 20мл). Органическую фазу сушили над CaCl2, после удаления растворителя получили частично кристаллизующееся масло желтого цвета. После перекристаллизации полученног продукта из дихлорметана получили 5.01 г твердого вещества бледно-желтого цвета (выход 42%). ЯМР 1Н (CDCl3, δ, м.д.): 10.32 (с, 1H, CHO). ЯМР 19F (CDCl3, δ, м.д.): -69.64 (с, 2F, CAr-Fмета); -48.62 (с, 6F, CF2); -45.65 (с, 6F, CF2); -45.26 (с, 6F, CF2); -44.56 (с, 6F, CF2); -43.07 (с, 6F, CF2); -3.29 (м, 9F, CF3).
2,4,6-три(пентафторфенилокси)-3,5-дифторбензальдегид (1f)
К раствору пентафторфенола (6.07 г, 33 ммоля) в сухом ДМФА (15мл) прибавляли пентафторбензальдегид (1.96 г, 10 ммолей) и KF (3.83 г, 66 ммолей) при интенсивном перемешивании. Реакционную смесь нагревали до 60°С и выдерживали при этой температуре 60 часов. Реакционную массу охлаждали, добавили 50мл диэтилового эфира, промыли насыщенным водным раствором NH4Cl (2 x 30мл) и водой (2 х 20мл). Органическую фазу сушили над CaCl2, после удаления растворителя получили частично кристаллизующееся масло желтого цвета. После перекристаллизации полученного продукта из дихлорметана 1.88 г твердого вещества бледно-желтого цвета (выход 27%). ЯМР 1Н (CDCl3, δ, м.д.): 10.48 (с, 1H, CHO). ЯМР 19F (CDCl3, δ, м.д.): -83.06 (т, 4F, J1 = 21.36 Гц, J2 = 18.31 Гц, орто-OC6F5(пара-F)); -79.64 (т, 1F, J 1= 21.36 Гц, J2 = 24.41 Гц, пара-OC6F5(пара-F)); -78.56 (д, 4F, J1 = 18.31 Гц, орто-OC6F5(орто-F)); -77.70 (д, 2F, J1 = 18.31 Гц, пара-OC6F5(орто-F)); -69.53 (с, 2F, C6F2CHO(мета-F)). Масс-спектр (EI-DIP), m/z: 520.0 [M-C6F5]+, 167.2 [C6F5]+.
мезо-тетракис(4-(2,2,3,3,3-пентафторпропил-1-окси)-2,3,5,6-тетрафторфенил)порфирин (2a)
В раствор 1a (1.2 г, 3.7 ммоля) и пиррола (0.25 г, 3.7 ммоля) в сухом дихлорметане (400 мл) пропускали аргон в течение 30 минут при перемешивании. Затем добавляли BF3xEt2O (0.14 мл, 1.1 ммоля) и перемешивали 21 час при комнатной температуре, добавляли DDQ (0.92 г, 4.07 ммоля) и кипятили 2 часа. После добавления Et3N (0.2 мл) растворитель удаляли, твердый остаток промывали холодным метанолом до бесцветного раствора. Полученный продукт очищали в 2 этапа: 1) флеш-хроматография (силикагель, хлористый метилен), 2) колоночная хроматография (силикагель, гексан – хлороформ = 1:1). Получено 0.32 г порошка фиолетового цвета (выход 23%). ЯМР 1Н (CDCl3, δ, м.д.): -2.89 (с, 2H, NH); 5.02 (т, 8H, J = 12.08 Гц), 8.96 (с, 8H, Pyr-H). ЯМР 19F (CDCl3, δ, м.д.): -78,55 (м, 8F, CAr-Fорто); -59.44 (м, 8F, CAr-Fмета); -46.55 (с, 8F, CF2); -5,52 (с, 12F, CF3). Масс-спектр (APCI), m/z: 1495.8 [M+H]+.
мезо-тетракис(4-(1,1-Н,Н-перфторпентил-1-окси)-2,3,5,6-тетрафторфенил)порфирин (2b)
В раствор 1b (1.97 г, 4.43 ммоля) и пиррола (0.29 г, 4.43 ммоля) в сухом дихлорметане (500мл) пропускали аргон в течение 30 минут при перемешивании. Затем добавляли BF3xEt2O (0.17 мл, 1.33 ммоля) и перемешивали 21 час при комнатной температуре, добавляли DDQ (1.11 г, 4.87 ммоля) и кипятили 2 часа. После добавления Et3N (0.24мл) растворитель удаляли, твердый остаток промывали холодным метанолом до бесцветного раствора. Полученный продукт очищали в 2 этапа: 1) флеш-хроматография (силикагель, хлористый метилен), 2) колоночная хроматография (силикагель, гексан - хлороформ=1:1). Получено 0.23 г порошка фиолетового цвета (выход 21%). ЯМР 1Н (CDCl3, δ, м.д.): -2.89 (с, 2H, NH); 5.07 (т, 8H, J = 12.87 Гц), 8.94 (с, 8H, Pyr-H). ЯМР 19F (CDCl3, δ, м.д.): -78.51 (м, 8F, CAr-Fорто); -59.44 (м, 8F, CAr-Fмета); -48.45 (с, 8F, CF2), -46.20 (с, 8F, CF2); -42.93 (с, 8F, CF2), -3.04 (с, 12F, CF3). Масс-спектр (APCI), m/z: 1895.4 [M+H]+.
мезо-тетракис(2,3,5,6-тетрафтор-4-(пентафторфенилокси)фенил)порфирин (2c)
В раствор 1c (0.72 г, 2 ммоля) и пиррола (0.18 г, 2,7 ммоля) в сухом дихлорметане (200мл) пропускали аргон в течение 30 минут при перемешивании. Затем добавили BF3xEt2O (0.1 мл, 0.8 ммоля) и перемешивали 20 часов при комнатной температуре, добавляли DDQ (0,61 г, 2,7 ммоля) и кипятили 2 часа. После охлаждения растворитель удаляли, полученный продукт очищали колоночной хроматографией (Al2O3, петролейный эфир – хлороформ = 6:1), продукт дополнительно промывали холодным метанолом. Получено 150мг порошка фиолетово-коричневого цвета (выход 18%). ЯМР 1Н (CDCl3, δ, м.д.): -2.88 (с, 2H, NH); 8.96 (с, 8H, Pyr-H). ЯМР 19F (CDCl3, δ, м.д.): -83.37 (т, 8F, J = 20.65 Гц, C6F5O (мета-F)); -80.95 (т, 4F, J 1= 22.95 Гц, J 2 = 20.65 Гц, C6F5O (пара-F)); -78.66 (д, 8F, J = 18.36 Гц, C6F4-Porph (мета-F)); -78.07 (д, 8F, J = 18.36 Гц, C6F5O (орто-F)); -58.80 (д, 8F, J = 13.77 Гц, C6F4-Porph (орто-F)). Вычислено для C68H10F36N4O4 (%): C, 50.08; H, 0.62; N, 3.44; F, 41.94. Найдено (%): C, 50.11; H, 0.49; N, 3.34; F, 41.65.
мезо-тетракис(3,5-дифтор-2,4,6-три(пентафторфенилокси)фенил)порфирин (2f)
В раствор 1f (0.68 г, 1 ммоль) и пиррола (0.06 г, 1 ммоль) в сухом хлороформе (100мл) с добавкой этанола (0.8 об. %) пропускали аргон в течение 30 минут при перемешивании. Затем добавляли BF3xEt2O (0.05 мл, 0.4 ммоля) и перемешивали 8 часов при комнатной температуре, добавляли DDQ (0.23 г, 1 ммоль) и перемешивали 1.5 часа. После добавления Et3N (0.05 мл) растворитель удаляли, твердый остаток промывали холодным метанолом до бесцветного раствора. Полученный продукт очищали в 2 этапа: 1) флеш-хроматография (силикагель, хлороформ), 2) колоночная хроматография (силикагель, гексан – хлороформ = 5:1). Получено 10мг порошка фиолетового цвета (выход 1%). ЯМР 1Н (CDCl3, δ, м.д.): -3.44 (с, 2H, NH); 9.01 (с, 8H, Pyr-H). ЯМР 19F (CDCl3, δ, м.д.): -85.65 (т, 16F, J1 = 18.31 Гц, J2 = 21.36 Гц, орто-OC6F5(мета-F)); -84.49 (т, 8F, J1 = 21.36 Гц, J2 = 18.31 Гц, пара-OC6F5(мета-F)); -83.27 (т, 8F, J1 = 18.31 Гц, орто-OC6F5(пара-F)); -81.14 (т, 4F, J = 21.36 Гц, пара-OC6F5(пара-F)); -79.80 (д, 16F, J = 18.31 Гц, орто-OC6F5(орто-F)), -78.55 (д, 8F, J = 21.36 Гц, пара-OC6F5(орто-F)); -71.09 (с, 8F, C6F2-Porph (мета-F)).
мезо-тетракис(2,3,5,6-тетрафтор-4-(1,1-Н,Н-перфторгептил-1-окси)фенил)порфирин (2g)
К раствору мезо-тетракис(пентафторфенил)порфирина (0.5 г, 0.51 ммоля) и 1,1-Н,Н-перфторгептанола-1 (2.16 г, 6.2 ммоль) в ТГФ (200 мл) прибавляли КОН (2.86 г, 51 ммоль) и кипятили в инертной атмосфере 4 часа. Упарили растворитель, остаток поместили в воду, экстрагировали диэтиловым эфиром. Органическую фазу промывали насыщенным водным раствором NaHCO3 (2 х 40мл) и сушили над CaCl2, После удаления растворителя полученную массу очищали колоночной хроматографией (силикагель, хлороформ). Получено 0.55 г порошка фиолетового цвета (выход 47%). ЯМР 1Н (CDCl3, δ, м.д.): -2.86 (с, 2H, NH), 5.11 (т, 8H, J1 = 12.79 Гц, J2 = 12.56 Гц, OCH2), 8.97 (с, 8H, Pyr-H). ЯМР 19F (CDCl3, δ, м.д.): -78.48 (м, 8F, CAr-Fорто), -59.44 (дд, 8F, J1 = 13.79Гц, J2 = 8.19 Гц, CAr-Fмета), -48.38 (м, 8F, CF2), -45.14 (м, 8F, CF2), -44.33 (м, 8F, CF2), -42.73 (м, 8F, CF2), -2.98 (т, 12F, J = 9.05 Гц, CF3). Вычислено для C72H18F68N4O4 (%): C, 35.00; H, 0.43; N, 2.00; F, 56.50. Найдено (%): C, 37.68; H, 0.79; N, 2.44; F, 56.30.
мезо-тетракис(2,3,5,6-тетрафтор-4-(1,1-Н,Н-перфторнонил-1-окси)фенил)порфирин (2h)
В раствор мезо-тетракис-(пентафторфенил)порфирина (0.1 г, 0.1 ммоль) в ТГФ (50мл) поместили 1,1-Н,Н-перфторнонанол-1 (1.2 г, 0.54 ммоля) и карбонат калия (1.38 г, 10 ммоля) и кипятили в инертной атмосфере 12 час, контролировали ход реакции по ТСХ (силикагель, гексан - хлороформ = 1:1). Из охлажденной реакционной смеси растворитель удалили, остаток поместили в воду, экстрагировали диэтиловым эфиром. Органическую фазу промывали насыщенным водным раствором NaHCO3, после осушки над CaCl2 растворитель удалили. Полученный остаток очищали флеш-хроматографией (окись алюминия, гексан - хлороформ = 1:1), затем колоночной хроматографией (окись алюминия, градиентное элюирование гексан - хлороформ). Получено 95 мг порошка фиолетового цвета (выход 35%). 1H ЯМР (CDCl3, δ, м.д.): -2.89 (с, 2H, NH), 5.06 (т, 8H, J = 12.72 Гц, OCH2), 8.94 (с, 8H, Pyr-H),. 19F ЯМР (CDCl3, δ, м.д.): -78.47 (м, 8F, CAr-Fорто), -59.43 (м, 8F, CAr-Fмета), -48.34 (с, 8F, CF2), -45.17 (с, 8F, CF2), -44.92 (с, 8F, CF2), -44.07 (с, 24F, CF2CF2CF2), -42.65 (с, 8F, CF2), -3.00 (с, 12F, CF3). Масс-спектр (APCI), m/z: 1834.6 [M+H-2C8F17CH2]+.
Список литературы
- DiMagno, S. G.; Dussault, P. H.; Schultz, J. A. J. Am. Chem. Soc. 1996, 118, 5312–5313.
- Pozzi, G.; Colombani, I.; Miglioli, M.; Montanari, F.; Quici, S. Tetrahedron 1997, 53, 6145–6162.
- Nakahara, H.; Liang, W.; Fukuda, K.; Wang, L.; Wada, T.; Sasabe, H. J. Colloid Interface Sci. 1998, 208, 14–22.
- Sun, H.; Smirnov, V. V; DiMagno, S. G. Inorg. Chem. 2003, 42, 6032–6040.
- Grancho, J. C. P.; Pereira, M. M.; Miguel, M. D. G.; Gonsalves, A. M. R.; Burrows, H. D. Photochem. Photobiol. 2002, 75, 249–256.
- Goll, J. G.; Moore, K. T.; Ghosh, A.; Therien, M. J. J. Am. Chem. Soc. 1996, 118, 8344–8354.
- Yi, W. Bin; Ma, J. J.; Jiang, L. Q.; Cai, C.; Zhang, W. J. Fluor. Chem. 2014, 157, 84–105.
- Gryko, D.; Wyrostek, D.; Nowak-Król, A.; Abramczyk, K.; Rogacki, M. Synthesis (Stuttg). 2008, 2008, 4028–4032.
- Lindsey, J. S.; Schreiman, I. C.; Hsu, H. C.; Kearney, P. C.; Marguerettaz, A. M. J. Org. Chem. 1987, 52, 827–836.
- Lindsey, J.; Wagner, R. J. Org. Chem. 1989, 828–836.
- Предварительные результаты данного исследования опубликованы в трудах - X. German-Russian-Ukrainian Symposium on Fluorine Chemistry, Berlin, November 26-28, 2014, P.19 (Belyaeva, E.; Sigan, A.; Gervits, L.; Chkanikov N.). Впоследствии оказалось, что одновременно с нами данный подход исследовался в работе немецких авторов - Golf, H. R.; Reissig, H.-U.; Wiehe, A. European J. Org. Chem. 2015, 2015, 1548–1568., которые рассмотрели взаимодействие TF20PPH2 с несколькими фторалифатическими спиртами.
- Figueira, A. C. B.; de Oliveira, K. T.; Serra, O. A. Dye. Pigment. 2011, 91, 383–388.
- Hyun, M. Y.; Jo, Y. D.; Lee, J. H.; Lee, H. G.; Park, H. M.; Hwang, I. H.; Kim, K. B.; Lee, S. J.; Kim, C. Chem. - A Eur. J. 2013, 19, 1810.
Статья рекомендована к публикации членом редколлегии д.х.н. проф. Н.Д. Чканиковым
Fluorine Notes, 2015, 102, 3-4


