Поступило в редакцию: март 2015
Fluorine Notes, 2015, 101, 3-4
Структурные свойства, Расчеты по теории функционала плотности (DFT), Обитали естественной связи (NBO) и энергии фторуглеродных соединений.
Shahriar Ghammamy, Farzane Yousefi
Department of Chemistry, Faculty of Science, Imam Khomeini International University, Qazvin,
Iran
e-mail: shghamamiii@yahoo.com или
farzaneyousefi@yahoo.com
Аннотация: Были теоретически изучены структурные и электронные свойства нового вещества со структурой C20H11F13ClN3O3S с помощью метода теории функционала плотности (1). Расчеты B3LYP/3-21G определили некоторое длинны выбранных связей и значение углов связей для C20H11F13ClN3O3S . В этой работе, с помощью метода теории функционала плотности с использованием стандартных базисных состояний 3-21G рассчитаны оптимизированные геометрии , частоты точек стационарности и траектории минимальной энергии двух новых веществ с формулой C20H11F13ClN3O3S. Детальная точечная группа соединения С1.
Ключевые слова:Электронная структура, Фторуглеродные соединения, расчеты по методу теории функционала плотности, колебательный анализ, уровень B3LYP.
1. Введение
Сульфамидные препараты до сих пор вместе с первыми лекарственными препаратами ( ампициллином и гентамицином) используются как химиопрепараты для лечения бактериальных инфекций человека, вызванных Escherichia coli. (2) Сульфонамиды это противомикробные препараты широко применяющиеся в животноводстве, и их остатки в пище могут представлять большой риск для здоровья людей. В молочной промышленности, ежедневно большое количество молока проверяется на присутствие сульфонамидов. Фторалканы могут использоваться как маслоотталкивающие, водооталктвающие вещества, фторполимеры, растворители, препараты для жидкостной вентиляции легких и сильные парниковые газы. Фторуглеродные жидкости бесцветны. Благодаря высокому молекулярному весу, они имеют высокую молекулярную плотность, почти вдвое выше воды. В последнее время отрицательное воздействие перфторированных веществ на окружающую среду вызывает особое беспокойство общественности, так как эти вещества широко распространены, устойчивы и их длиноцепные гомологи способны к накоплению в живых тканях. Во время нашей работы, мы описали расчеты оптимизированных геометрий, распределения и электронных структур для этого соединения. Структура соединения была оптимизирована с помощью метода теории функционала плотности ( B3LYP) с использованием стандартных базисных состояний 3-21G по программе Gaussian 09. Метод теории функциональной плотности был использован для определения оптимальной структуры C20H11F13ClN3O3S. Первоначальные расчеты были проведены на уровне Метода теории функциональной плотности с валентно-расщепленными поляризационными базисами 3-21G.
2. Экспериментальная часть
2.1 Вещества и реагенты
Оптимизированные структурные параметры были использованы при вычислении колебательной чистоты на уровне DFT для характеристики минимальных значений стационарных точек. Все вычисления выполнены с помощью программы Gaussian 09. Гармоничные вибрационные частоты (ν) в см -1 и интенсивности инфракрасного излучения ( int) в километрах на моль всех веществ были изучены на одном уровне на соответствующих полностью оптимизированных геометрий. Энергетический минимум молекулярных геометрий был обнаружен по методу минимизации энергии, в соответствии со всеми геометрическими координатами без налагания симметрических ограничений.
2.2 Изучение орбиталей естественных связей (NBO) структур
Рассчитанные гибридизации NBO являются важными параметрами нашего исследования. Структура соединения была оптимизирована с помощью метода теории функционала плотности (B3LYP) с использованием стандартных базисных состояний 3-21G по программе Gaussian 09. Методы теории функционала плотности были применены для оптимизации структур C20H11F13ClN3O3S.
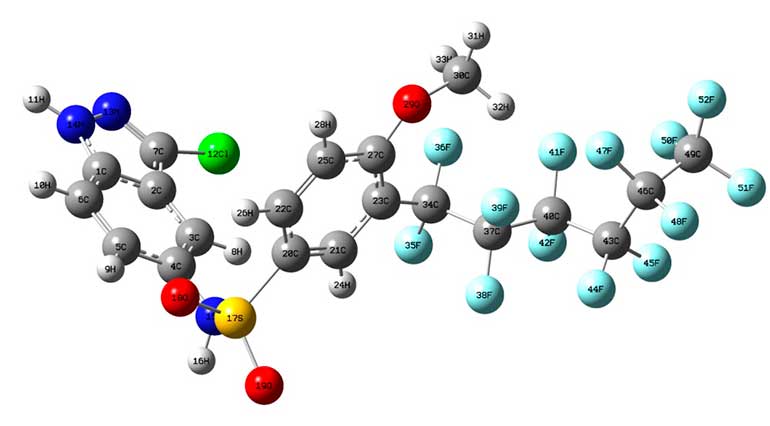
Рисунок 1. Оптимизированные геометрии C20H11F13ClN3O3S на уровне теории B3LYP/3-21
Таблица 1. Геометрические параметры оптимизированные для C20H11F13ClN3O3S некоторых выбранных длин связей (Å) и углов (◦).
|
|
Длина связи |
|
Длина связи |
|
Длина связи |
|
C7-Cl12 |
1.7959 |
H31-F52 |
0.9053 |
C43-F44 |
1.35 |
|
N13-N14 |
1.4114 |
H33-F39 |
1.3936 |
C43-F45 |
1.35 |
|
N15-S17 |
1.7765 |
C34-F35 |
1.35 |
C46-F47 |
1.35 |
|
S17-O18 |
105992 |
C37-F39 |
1.35 |
C46-F48 |
1.35 |
|
S17-O19 |
1.5874 |
C40-F42 |
1.35 |
C49-F50 |
1.35 |
|
O29-C30 |
1.4639 |
F41-F52 |
1.717 |
C49-F51 |
1.35 |
|
C30-F52 |
1.4049 |
F41-F50 |
1.4323 |
F41-C49 |
1.7674 |
|
|
Угол |
|
Угол |
|
Угол |
|
N15-S17-O19 |
102.3412 |
F35-C34-F36 |
109.4712 |
H32-C46-F48 |
150.7584 |
|
C2-C7-Cl12 |
124.3832 |
F38 |
109.4712 |
F47-C46-F48 |
109.4713 |
|
O29-C30-F52 |
111.7984 |
F41-C40-F42 |
109.4712 |
F41-C49-F51 |
151.1907 |
|
Cl12-C7-N13 |
121.5988 |
C40-F41-F50 |
108.903 |
F50-C49-F52 |
109.4713 |
|
C7-N13-N14 |
103.8277 |
C40-F41-F52 |
124.9545 |
F51-C49-F52 |
109.4712 |
|
C30-H32-F47 |
134.5953 |
F50-F41-F52 |
88.3745 |
C30-F52-F41 |
73.4312 |
|
N15-S17-O18 |
114.0423 |
F44-C43-F45 |
109.4712 |
H31-F52-F41 |
51.3647 |
Орбитали естественных связей (NBO) это локализованные орбитали с несколькими центрами, которые описывают, молекулярную связующую модель пар электронов по Льюису в оптимально компактной форме. Точнее, NBO это ортонормальное множество “локализованных “ максимальной занятости” орбиталей, чьи ведущие члены N/2 (N члены в незамкнутой оболочке) дают самое точное возможное описание общей N-электронной плотности типа Льюиса. Анализ проведен посредством изучения всех возможных взаимодействий между “ заполненными” (донорными) NBO льюисовского типа и “ пустыми” (акцепторными) NBO нельюисовского типа, и с помощью оценки их энергетической значимости теорией возмущений второго порядка. Так как эти взаимодействия ведут к передаче занятости от локализованных NBO идеализированной структуру Льюиса пустым нельюисовским орбиталям ( Следовательно речь идет об описании передачи от идеальной структуры Льюиса), они называются коррекцией “делокализации” естественной структуры Льюиса нулевого порядка.
Натуральные заряды были вычислены с помощью модуля орбиталей естественных связей в программе Gaussian 09W. Эти количества получены согласно анализу популяций NBO. Модель представляет картину орбитали ближе к классической структуре Льюиса. Анализ NBO включает гибридизацию избранных связей рассчитанных методом B3LYP с использованием стандартных базисных состояний 3-21G ( таблицы 2 и 3).
Таблица 2. Рассчитанные гибридизации NBO для C20H11F13ClN3O3S при помощи B3LYP/3-21G.
|
B3LYP |
Атом |
Связь |
B3LYP |
Атом |
Связь |
|
S1P4.55,S1P2.73 |
N13-N14 |
N-N |
S1P2.83,S1P6.03 |
C7-Cl12 |
C-Cl |
|
S1P2.22,S1P8.76 |
S17-O18 |
S-O |
S1P4.51,S1P4.18 |
N15-S17 |
N-S |
|
S1P4.19,S1P3.18 |
C4-F35 |
C-F |
S1P2.58,S1P8.76 |
S17-O19 |
S-O |
|
S1P2.59,S1P4.76 |
O29-C30 |
O-C |
S1P2.57,S1 |
N15-H16 |
N-H |
|
S1P3.98,S1P3.12 |
C37-F38 |
C-F |
S1P4.16,S1P3.16 |
C34-F36 |
C-F |
|
S1P4.07,S1P3.23 |
C40-F41 |
C-F |
S1P4.19,S1P3.22 |
C37-F39 |
C-F |
|
S1P3.97,S1P3.19 |
C43-F44 |
C-F |
S1P4.00,S1P3.14 |
C40-F42 |
C-F |
Эти данные показывают, что существует гиперсопряжение электронов между лигандами атомов с центральным атомом металла. Данные сопряжения основываются на p-d π – связях. Рассчитанные для C20H11F13ClN3O3S гибридизации NBO показывают, что все соединения имеют SPX гибридизацию и неплоскую конфигурацию. Тотальная гибридизацию этих молекул это SPX , что подтверждено их структурой. Количество гибридизированных связей показало равнозначность между углами центральных атомов (таблица 2) показывающими искажение нормальных VSEPR структур и подтверждающими девиацию от VSEPR структур. (Рисунок 2)
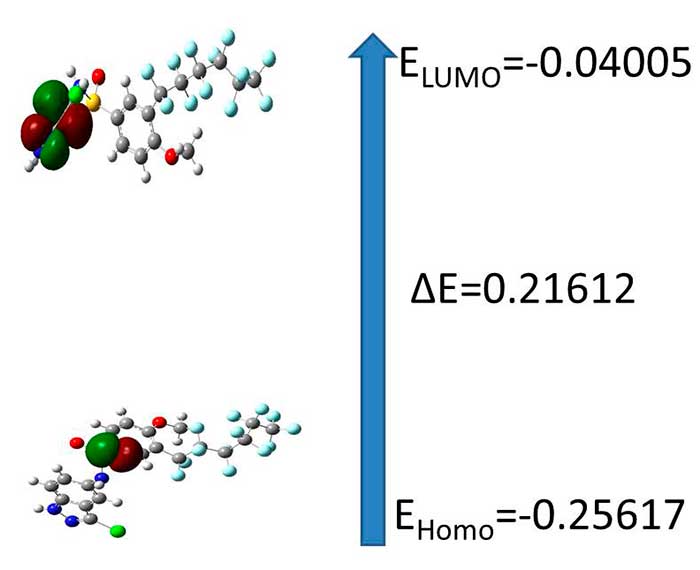
Рисунок 2. Атомная орбиталь граничной молекулярной орбитали для C20H11F13ClN3O3S при помощи B3LYP/3-21G
Были рассчитаны и подтверждены с опубликованными теоретическими данными некоторые термодинамические параметры частот для C20H11F13ClN3O3S , энергия нулевых колебаний, Энергия коррекции, Энтальпии , свободная энергия Гибса
Таблица 3. Некоторые термодинамические параметры частот для C20H11F13ClN3O3S , энергия нулевых колебаний, Энергия коррекции, Энтальпии , свободная энергия Гибса.
|
C20H11F13ClN3O3S |
|
Коррекция нулевых колебаний = 0.314221 (Hartree/Particle) |
|
Тепловая коррекция к энергии = 0.351115 |
|
Тепловая коррекция к энтальпии = 0.352059 |
|
Тепловая коррекция к свободной энергии Гибса = 0.240518 |
2.3 Электронная плотность
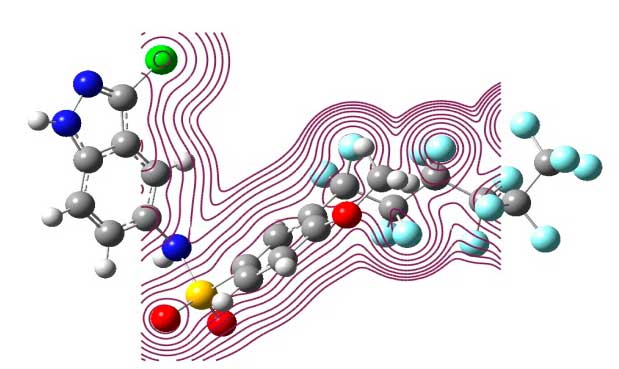
Рисунок 3. Тотальная электронная плотность поверхности C20H11F13ClN3O3S
2.4 Граничная молекулярная орбиталь
HOMO (ВЗМО высшая заселенная молекулярная орбиталь) представляет способность отдавать электрон, LUMO (низшая незанятая молекулярная орбиталь), как акцептор электрона представляет способность принимать электрон. Энергии HOMO и LUMO были рассчитаны по методу B3LYP/3-21G. Эта электронная абсорбция относиться к переходу от начального к первому возбужденному состоянию и в основном описывается возбуждением одного электрона с высшей заселенной молекулярной орбитали и орбитали (LUMO). Как высшая заселенная молекулярная орбиталь (HOMO) так и низшая незанятая молекулярная орбиталь являются главными орбиталями, принимающими участие в химической стабильности. Так как энергия HOMO напрямую связана с ионизационным потенциалом, Энергия LUMO напрямую связана со сродством к электрону. Энергетическая разница орбиталей HOMO и LUMO называется энергетическим интервалом, что является важным для стабильности структур. В дополнение, на рисунке 2 представлены 3D модели Высших заселенных молекулярных орбиталей (HOMO) и низших незянятых молекулярных орбиталей (LUMO). Энергии HOMO-LUMO были также рассчитаны ) с использованием стандартных базисных состояний 3-21G и полученные данные соответственно указаны на рисунке.
ИК спектр
Инфракрасные спектры могут быть получены у образцов во всех фазах (жидкой, твердой, газообразной) Жидкости обычно анализируются, нанесением тонкой пленки, между двумя полированными соляными пластинами (обратите внимание, что стекло поглощает инфракрасное излучение, в то время как NaCl остается прозрачным). Если для растворения твердых веществ используются растворители, необходимо избегать затемнения важных частей спектра, поглощением растворителя. Обычно используются перхлоророванные растворители, такие как тетрахлорметан, хлороформ и тетрахлорэтен. В качестве альтернативы, твердые вещества, также можно нанести на тонкую пластину KBr, под высоким давлением, или смешиваются с небольшим количеством нелетучей жидкости до пасты, которая размазывается между соляными пластинами. Однако в этой работе мы получили теоретический ИК спектр с помощью программы Gaussian 09. Частоты функциональных групп представлены на рисунке 4.
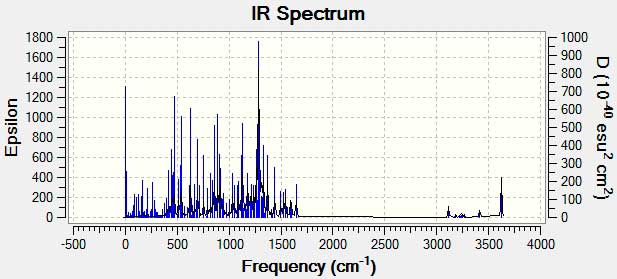
Рисунок 4. ИК спектр для С20H11F13ClN3O3S
ЯМР спектр
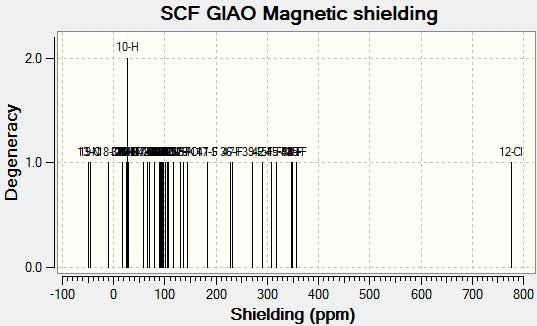
Рисунок 5. ЯМР спектр для C20H11F13ClN3O3S
Заключение
В данной статье нас интересовало теоретическое изучение выбранного фторуглеродного соединения. С помощью методов теории функциональной плотности (B3LYP) c базисным набором 3-21G были вычислены оптимизированные геометрии, частоты стационарной точки и пути минимальной энергии. Расчеты B3LYP/3-21G определили некоторые избранные длины связей и величины углов для С20H11F13ClN3O3S. Точечная группа соединения это С1.
Благодарности
Выражаем глубокую благодарность за финансовую поддержку Научному Совету Международного университета имени Имама Хомейни и предоставление Гранта N 751387-91 и техническую поддержку Университета Тарбиат Муаллем.
Список литературы
- Ghadah S. Alghamdi and Ali. Z. Alzahrani, Bonding Formation and Orbitals Nature of Zno Structure .2013.13.9.751
- Monika, W. A.; Siddique, A. D. Portugaliae ElectrochimActa 2005, 23, 445.
- O'Hagan, D . February 2008. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 37 (2): 308–19
- Sudha. S,Sundaraganesan.N,Kurt,Cinar.M. 2011 Karabacak, Molecularstructure, vibrational spectroscopic, first order hyperpolarizability and HOMO, LUMO studies of 2-aminobenzimidazole.Journal of Molecular Structure,84: 184-195.
- Lemal, D. M. 2004. Perspective on fluorocarbon c?hemistry. Journal of Organic Chemistry, 69 (1): 1–11.
- Murphy, C.D, Schaffrath, C. O'Hagan D2003Fluorinated natural products: the biosynthesis of fluoroacetate and 4-fluorothreonine in Streptomyces cattleya" Chemosphere,52(2):455-61.
- Lewandowski ,G. Meissner E, Milchert E.Hazard ,J. 2006. Special applications of fluorinated organic compounds.136(3):385–91.
- Becke, A. D. 1993 Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys., 98: 5648-5652
- Sundaraganesan, N. and S. Ilakiamani, 2007. Dominic Joshua B Vibrational spectroscopy investigation using ab initio and density functional theory analysis on the structure of 3, 4-dimethylbenzaldehyde. Spectrochimica Acta Part A., 68: 680-687
- Lewis, D. F. V., C. Ioannides, and Parke, D. V. 1994. Interaction of a series of nitriles with the alcohol-inducible isoform of P450: computer analysis of structure- activity relationships. Xenobiotica, 24: 401-408.
- Ralph, G. 1992. Chemical hardness and the electronic chemical potential Inorganic, chimica Acta, 198: 781-786.
- Zhang, W.Curran D.P. 2006. Synthetic Application of Fluorous. Tetrahedron 62: 11837–11865.
- Smith, M. C., Ciao.Y, Wang ,H. andGeorge, S. J. 2005. Coucouvanis D., Koutmos M, Sturhahn W, Alp EA, Zhao J, Kramer SP Normal-Mode Analysis of FeCl4- and Fe2S2Cl42- via Vibrational Mossbauer, Resonance Raman, and FT-IR Spectroscopies.Inorg. Chem., 44: 5562-5570.
- Vrajmasu, V. V., Mu¨nck,E. and E. L. Bominaar, 2004. Theoretical Analysis of the Jahn−Teller Distortions in Tetrathiolato Iron(II) Complexes. Inorg. Chem., 43: 4862–4866.
- Ghammamy, Sh., K. Mehrani,Rostamzadehmansor, S. and Sahebalzamani,H. 2011. Density functional theory studies on the structure, vibrational spectra of three new tetrahalogenoferrate (III) complexes. Natural Science, 3, 683-688.
- Frisch, M. J. Trucks, G. W. 1998. GASSIAN 98 (Revision A. 3) Gaussian Inc., Pittsburgh, PA, USA.
Статья рекомендована к публикации членом редколлегии д.х.н. проф. С.М. Игумновым
Fluorine Notes, 2015, 101, 3-4


