Поступило в редакцию: ноябрь 2014
УДК 547.321;547.752;547.82
Fluorine Notes, 2014, 97, 3-4
Синтез 4-фторалкил-9Н-пиримидо[4,5-b]индолов
А.С. Голубев*, О.А. Митюшина, А.Ф. Шидловский, К.Ю. Супоницкий, Н.Д. Чкаников
Институт элементоорганических соединений им. А.Н. Несмеянова Российской академии наук, Российская Федерация,
119991 Москва, ул. Вавилова, 28.
e-mail: golubev@ineos.ac.ru
Факс: (499) 135 5085.
Аннотация. Разработан метод синтеза 2-метилтио-4-фторалкил-9Н-пиримидо[4,5-b]индолов 1 исходя из легкодоступных 2-метилтио-6-фторалкил-пиримидинон-4(3Н)-онов 2. Ключевой стадией синтеза является внутримолекулярная циклизация по Хеку 4-(анилино)-5-иодо-2-метилтио-6-(фторалкил)пиримидинов 5.
Ключевые слова: 9Н-пиримидо[4,5-b]индолы, 2-метилтио-4-фторалкил-9Н-пиримидо[4,5-b]индолы, внутримолекулярная циклизация по Хеку, 2-метилтио-6-(фторалкил)пиримидинон-4(3Н)-оны
9Н-Пиримидо[4,5-b]индолы – важный класс гетероциклических соединений. Первые представители этой гетероциклической системы были синтезированы во ВНИХФИ О.Ю. Магидсоном и Р.Г. Глушковым [1].
9Н-Пиримидо[4,5-b]индолы проявляют широкий спектр биологической активности [2-7]. В ряду этих гетероциклов ведется активный поиск новых противоопухолевых препаратов. 9Н-Пиримидо[4,5-b]индолы были использованы [7] в дизайне противоопухолевых препаратов, мишенью которых являлся белок тубулин и построенные из него микротрубочки [8]. Авторы работы [7] подтвердили важность присутствия в конструируемом ингибиторе полимеризации тубулина набора определенных фармакофорных признаков: жесткого остова молекулы, наличие гидрофобного фрагмента в положении 4 трицикла, на ядре С функциональных групп - акцепторов водородной связи, а на ядре А функциональных групп – доноров водородной связи.
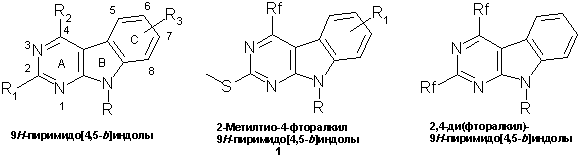
Введение фторалкильной группы в молекулу конструируемого ингибитора – распространенный прием обеспечения взаимодействия молекулы-ингибитора с гидрофобными карманами ингибируемого энзима [9].
Целью данной работы явился синтез 4-фторалкил-2-метилтио-9Н-пиримидо[4,5-b]индолов (1), производные которых могут являться ингибиторами полимеризации тубулина. Известные из литературы 2,4-ди(фторалкил)-9Н-пиримидо[4,5-b]индолы, полученные обращенной реакцией Дильса-Альдера (IDA реакцией) 2-аминоиндола с 2,4,6-трис(фторалкил)-1,3,5-триазинами [10,11], имеют определенные ограничения с точки зрения дизайна ингибиторов, поскольку варьирование природы заместителя в положении 2 трицикла в рамках данного синтетического подхода не представляется возможным. Использование же 2-алкилтио-заместителей в пиримидинах является обычным синтетическим подходом при получении различных С2-замещенных пиримидинов [12].
9Н-Пиримидо[4,5-b]индолы могут быть синтезированы исходя из производных индола [13a,b] или из пиримидинов [13с]. Используя последний подход, необходимо учесть, что фторалкильные группы являются сильными электроноакцепторными группами, в значительной степени дезактивирующими пиримидиновый цикл. Этот фактор скорее всего затруднил бы реакции получения 9Н-пиримидо[4,5-b]индолов, протекание которых определяется донорностью пиримидинового ядра, в частности, реакцию Неницеску [14] или варианты синтеза индолов по Фишеру [7,15] и Бишлеру [2,16]. В то же время палладий-катализируемые реакции арилбромидов или арилиодидов (различные кросс-сочетания, реакция Хека) протекают легче при наличии сильной электроноакцепторной группы в ароматическом ядре [17]. Ранее 5-иод-4-(анилино)пиримидины были использованы в синтезе 4-незамещенных 9Н-пиримидо[4,5-b]индолов в условиях внутримолекулярной циклизации по Хеку [18] и кросс-сочетания по Стилле [19]. Мы полагали, что внутримолекулярная циклизация по Хеку может быть успешно использована в синтезе 4-замещенных 9Н-пиримидо[4,5-b]индолов.
Обсуждение результатов
Нами разработан 4-х-стадийный метод синтеза 2-метилтио-4-фторалкил-9Н-пиримидо[4,5-b]индолов (1) исходя из легкодоступных 2-метилтио-6-фторалкил-пиримидинон-4(3Н)-онов (2) (схема 1). Синтетическая схема включает цепочку последовательных превращений: иодирование соединений (2) по положению 5 пиримидинового цикла с образованием иодпроизводных (3), хлорирование соединений (3) с получением хлорпиримидинов (4), аминирование последних анилинами с образованием 4-анилино-5-иодпиримидинов (5). Ключевой стадией всей синтетической схемы является внутримолекулярная циклизация по Хеку 4-анилино-2-метилтио-5-иодо-6-(фторалкил)пиримидинов (5). Фторалкильные заместители, рассматриваемые в рамках данной работы, представляли собой трифторметильную CF3- и дифторметильную CF2H- группы.
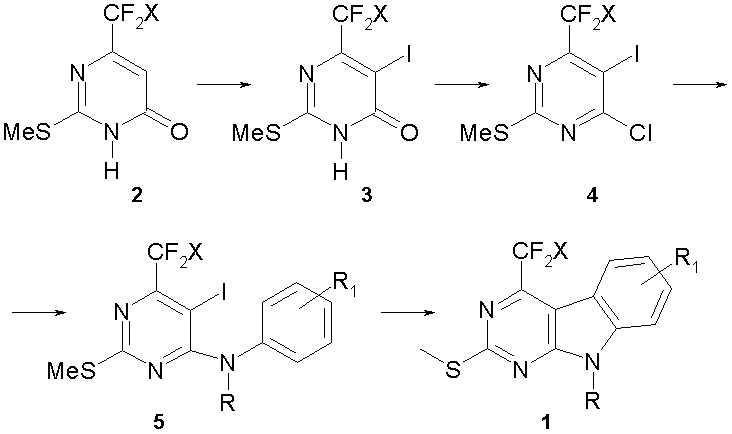
Схема 1
2-Метилтио-6-фторалкил-пиримидинон-4(3Н)-оны (2) – либо коммерчески доступны (в случае 6-СF3-аналога (2а)) либо могут быть легко получены циклоконденсацией бидентантного нуклеофила – гемисульфата S-метилизотиомочевины с трифтор- и дифторацетоуксусным эфирами (схема 2). Наилучшие выходы соединений (2a,b) были достигнуты при использовании 10%-ного водного раствора NaOH [20].
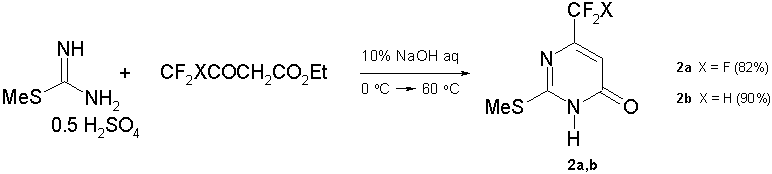
Схема 2
Стандартная процедура С5-иодирования пиримидонов – реакция с иодом в водном растворе NaOH [21] в случае пиримидинонов (2a,b) привела к неудовлетворительным результатам. Оптимальной оказалась реакция с N-иодсукцинимидом в кипящем ацетонитриле (схема 3) [22]. 5-Иодпиримидоны (3a,b) были выделены с выходом 92% и 82% соответственно.
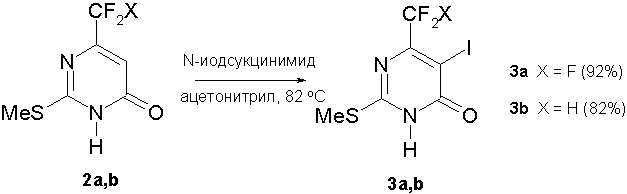
Схема 3
Несколько меньший выход дифторметил-аналога (3b) по сравнению с трифторметил-аналогом (3а) объясняется потерями при очистке продукта (3b). Для отделения продуктов (3a,b) от сукцинимида их нагревали в воде при 50°С. При этом выход дифторметил-аналога (3b) заметно упал из-за его частичной растворимости в воде при 50°С. В то время как более гидрофобный характер CF3-группы приводит к незначительной растворимости в воде трифторметил-аналога (3а) и, как следствие, к меньшим потерям в процессе очистки.
При кипячении пиримидинонов (3a,b) в хлорокиси фосфора POCl3 без добавки третичного амина мы наблюдали их крайне низкую конверсию в целевые хлорпиримидины (4a,b). Стандартная процедура хлорирования пиримидинонов – кипячение в хлорокиси фосфора в присутствии N,N-диметиланилина [23] в случае пиримидинонов (3a,b) привела к неудовлетворительным результатам. Хлорпиримидины (4a,b), содержащие в качестве примесей продукты их вторичной реакции с N,N-диметиланилином, требовали дополнительной очистки колоночной хроматографией. Наилучшие результаты были достигнуты при использовании комбинации POCl3 – ДБУ (1,8-диазабицикло[5.4.0]ундец-7-ен). В присутствии 50-60 мол % ДБУ реакция хлорирования протекает однозначно, приводя исключительно к хлорпиримидинам (4a,b) с высокими выходами (схема 4).
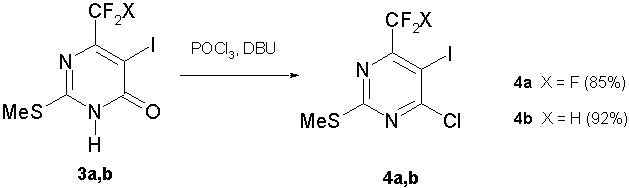
Схема 4
Синтез 4-(анилино)пиримидинов (5a-с) осуществляли кипячением в диоксане хлорпиримидинов (4a,b) и небольшого избытка (15-20 мол %) соответствующего анилина в присутствии эквимольного количества N,N-диизопропилэтиламина (основания Хюнига) и 1-2 эквивалентов LiBr (схема 5).
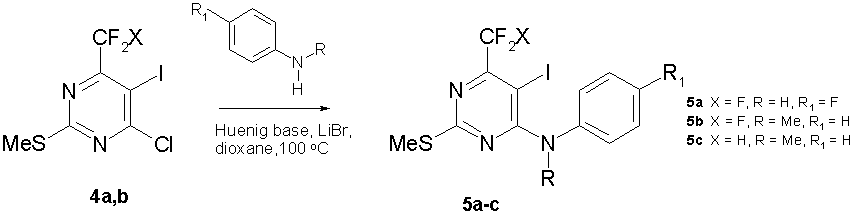
Схема 5
Добавка LiBr в реакционную массу позволила резко сократить время и увеличить выходы реакции получения пиримидинов (5a-с).
Внутримолекулярную циклизацию по Хеку проводили нагреванием смеси 4-(анилино)пиримидина (5) (1 экв), Pd(OAc)2 (10 моль %), трициклогексилфосфина PCy3 (20 моль %) и ацетата натрия (2 экв) в диметилформамиде при 125 °С [18]. В случае соединения (5a) после 4 ч нагрева наблюдали его полную конверсию и образование двух продуктов реакции. Целевой пиримидо[4,5-b]индол (1а) был выделен с выходом 25% (cхема 6). Основным продуктом реакции с выходом 60% оказался продукт деиодирования (6а). Все попытки увеличить выход целевого соединения (1а) оказались безуспешными. Так, заменив лиганд PCy3 на трифенилфосфин, мы получили примерно такой же низкий выход соединения (1а). Использование в качестве оснований триэтиламина или фторида калия также существенно не изменили ситуацию. Безрезультатно окончились попытки увеличить концентрацию раствора исходного до концентрации 0.5 М и выше. Безлигандный вариант (протокол Джеффери) был рекомендован для проведения циклизаций по Хеку в ряду азотсодержащих гетероциклов [24]. Однако, нами зафиксировано полное отсутствие целевого пиримидо[4,5-b]индола (1а) при нагревании смеси соединения (5a) (1 экв), Pd(OAc)2 (10 моль %), тетрабутиламмоний бромида (1 экв) и карбоната калия (3 экв) в ДМФА при 125 °С.
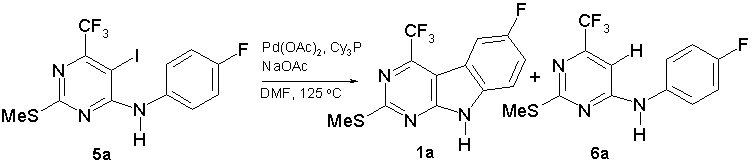
Схема 6
В то же время при нагревании N-метилированных аналогов (5b,с) в присутствии Pd(OAc)2 (10 моль%), PCy3 (20 моль%), ацетата натрия (2 экв) в ДМФА при 125°С целевые пиримидо[4,5-b]индолы (1b,c) были выделены с препаративными выходами (cхема 7).
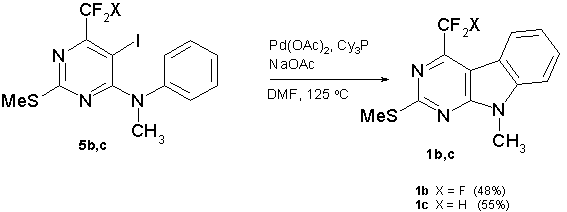
Схема 7
Строение целевых пиримидо[4,5-b]индолов (1а-c) подтверждено данными ЯМР спектроскопии, масс-спектрометрии и элементным анализом, а в случае соединения (1с) данными рентгеноструктурного анализа. Общий вид молекулы соединения (1с) и принятая в структурном эксперименте нумерация атомов приводятся на рис.1.
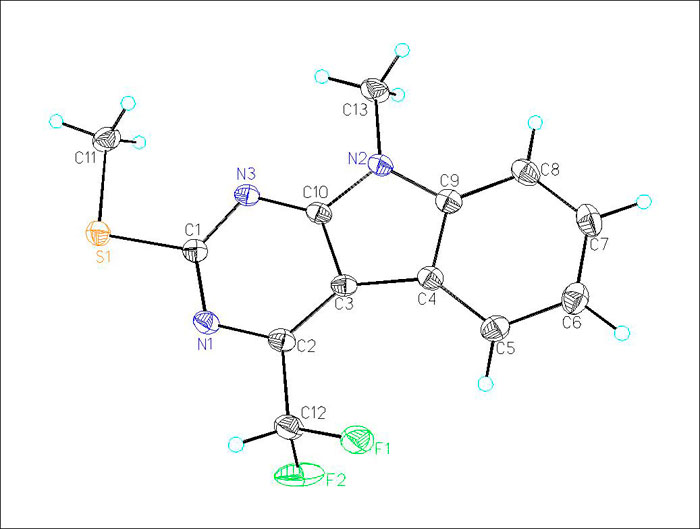
Рис. 1. Общий вид молекулы соединения 1с по результатам РСА в тепловых эллипсоидах 50% вероятности.
Экспериментальная часть
Спектры ЯМР 1Н и 13С{1H} были зарегистрированы на спектрометре Bruker AvanceTM300 (300.13 MHz) или на спектрометре «Bruker AvanceTM 600» (600.22 MHz). Спектры ЯМР 13С{1H} были зарегистрированы на спектрометре «Bruker AvanceTM 600» с рабочей частотой по ядрам 13С 150.925 MГц. Спектры ЯМР 19F {1H} были зарегистрированы на спектрометре «Bruker AvanceTM 300» с рабочей частотой по ядрам 19F 288.38 MГц. Химические сдвиги ядер 19F определяли относительно трифторуксусной кислоты как внешнего стандарта.
Масс-спектры получены на приборе Finnigan Polaris Q (ионная ловушка), энергия ионизирующего излучения 70 эВ, методика ввода образца DIP. Для колоночной хроматографии использован силикагель с размером частиц 0.06-0.20 мм (Merck Kieselgel 60). Обнаружение веществ осуществляли, используя пластинки ТСХ (Merck Kieselgel 60 F254). N,N-Диметилформамид («Catrosa») дополнительно очищали перегонкой в вакууме над СаН2. Этил-4,4,4-трифторацетоацетат был приобретен у фирмы «P&M», этил-4,4-дифторацетоацетат у фирмы «Aldrich». Элементный анализ выполнен в лаборатории элементного анализа ИНЭОС РАН. Растворители: петролейный эфир (ПЭ), этилацетат (ЭА) и N,N-диметилформамид (ДМФА).
2-Метилтио-6-(трифторметил)пиримидин-4(3Н)-он (2a).
К раствору гемисульфата S-метилизотиомочевины (7.0 г, 50 ммоль) в 40 мл 10% NaOH при 0 °С прибавили по каплям этил-4,4,4-трифторацетоацетат (9.2 г, 50 ммоль). Реакционную смесь перемешивали 2 часа при 0-5 °С, отогрели до 20 °С, нагревали при 65 °С в течение 1 часа, охладили до 20 °С и вылили на воду. Полученный раствор подкислили до рН 6 уксусной кислотой. Выпавший осадок отфильтровали, промыли многократно водой и сушили на воздухе. Получено 8.6 г (82%) соединения (2a) в виде белых кристаллов. Т.пл. 178-179 oC (см. лит.[23]: 179-180 °С). 1H NMR (600.22 MHz, CDCl3): δ 6.64 (1H, с, H5), 2.70 (3H, с, S-Me). 13С NMR (150.93 MHz, d6-DMSO): δ 166.6, 163.0, 151.5 (кв, 2JC,F= 34 Hz, C6-CF3), 121.0 (кв, 1JC,F= 274 Hz, CF3), 108.0, 13.4. 19F NMR (282.38 MHz, CDCl3): δ 5.85 (с, CF3). EIMS 70 eV, m/z: 211 [M + H]+ (11), 210 [M]+ (100), 191 [M – F]+ (10), 190 [M – НF]+ (56), 163 [M – SMe]+ (48), 162 [M – H–SMe]+ (24), 141 [M – CF3]+ (22).
6-Дифторметил-2-(метилтио)пиримидин-4(3Н)-он (2b).
Соединение (2b) получено аналогично исходя из гемисульфата S-метилтиомочевины и этил-4,4-дифторацетоацетата в виде белых кристаллов. Выход 90%. Т.пл. 201-202 oC. 1H NMR (300.13 MHz, d6-DMSO): δ 6.88 (1H, т, J = 54 Hz, CF2H), 6.35 (1H, с, H5), 2.50 (3H, с, S-Me). 13С NMR (150.93 MHz, d6-DMSO): δ 165.7, 163.5, 157.1 (т, 2JC,F= 23 Hz, C6-CF2H), 112.5 (т, 1JC,F= 237 Hz, CF2H), 107.2, 13.3. 19F NMR (282.38 MHz, d6-DMSO): δ -45.06 (с, CF2H). EIMS 70 eV, m/z: 193 [M + H]+ (12), 192 [M]+ (100), 191 [M– H]+ (56), 172 [M – HF]+ (65), 145 [M – SMe]+ (27), 144 [M – H – SMe]+ (29), 141 [M – CF2H]+ (46). Вычислено для C6H6F2N2OS: C, 37.50; H, 3.15; N, 14.58. Найдено C, 37.33; H, 3.17; N, 14.44.
5-Иод-2-метилтио-6-(трифторметил)пиримидин-4(3Н)-он (3a).
К смеси соединения (2a) (3.15 г, 15 ммоль) и N-иодсукцинимида (3.82 г, 17 ммоль) добавили ацетонитрил (40 мл). Полученную суспензию кипятили в течение 2 часов и охладили до 20 °С. Ацетонитрил отогнали в вакууме, остаток растворили в этилацетате (50 мл) и обработали 5% раствором Na2S2O3 до обесцвечивания органической фазы. Органический слой отделили и упарили в вакууме. К остатку (4.8 г кремовых кристаллов) добавили 50 мл воды. Полученную суспензию нагревали при 50 °С в течение 30 мин, охладили, отфильтровали и сушили на воздухе. Получено 4.63 г (92%) соединения (3a) в виде кремовых кристаллов. Т.пл. 241-243 oC (этанол-вода). 1H NMR (300.13 MHz, d4-метанол): δ 2.65 (3H, с, S-Me). 13С NMR (150.93 MHz, d4-метанол): δ 163.8, 161.4, 154.3 (кв, 2JC,F= 33 Hz, C6-CF3), 120.5 (кв, 1JC,F= 276 Hz, CF3), 82.8. 12.2. 19F NMR (282.38 MHz, d4-метанол): δ 9.93 (с, CF3). EIMS 70 eV, m/z: 337 [M + H]+ (15), 336 [M]+ (84), 289 [M – SMe]+ (8), 210 [M + H – I]+ (16), 209 [M – I]+ (100). Вычислено для C6H4F3IN2OS: C, 21.44; H, 1.20; N, 8.34. Найдено C, 21.22; H, 0.98; N, 8.21.
6-Дифторметил-5-иод-2-(метилтио)пиримидин-4(3Н)-он (3b).
Соединение (3b) получено аналогично из соединения (2b) в виде кремовых кристаллов. Выход 82%. Т.пл. 249-250 oC (этанол-вода). 1H NMR (300.13 MHz, d6-DMSO): δ 11.13 (1Н, уш.с, NН), 6.95 (1H, т, J = 53 Hz, CF2H), 2.61 (3H, с, S-Me). 1H NMR (300.13 MHz, CDCl3): δ 10.81 (1Н, уш.с, NН), 6.79 (1H, т, J = 53 Hz, CF2H), 2.71 (3H, с, S-Me). 13С NMR (150.93 MHz, d6-DMSO): δ 163.9, 160.8, 156.4 (т, 2JC,F= 21Hz, C6-CF2H), 114.9 (т, 1JC,F= 242Hz, CF3), 87.2. 13.5. 19F NMR (282.38 MHz, d6-DMSO): δ -42.67 (с, CF3). EIMS 70 eV, m/z: 319 [M + H]+ (9), 318 [M]+ (82), 271 [M – SMe]+ (11), 192 [M + H – I]+ (9), 191 [M – I]+ (100). Вычислено для C6H5F2IN2OS: C, 22.66; H, 1.58; N, 8.81. Найдено C, 22.23; H, 1.43; N, 8.65.
5-Иод-2-метилтио-6-трифторметил-4-хлорпиримидин (4а).
К соединению (3a) (2.85 г, 8.5 ммоль) добавили хлорокись фосфора (20 мл). К полученной желтой суспензии при охлаждении до 10 °С осторожно по каплям добавили ДБУ (0.76 г, 5 ммоль). Реакционную смесь кипятили до прекращения выделения HCl. По окончании реакции (время реакции 1.5 часа) хлорокись фосфора отогнали в вакууме. Остаток обработали ледяной водой, экстрагировали хлористым метиленом (2 х 30 мл). Органическую фазу промыли 5% раствором NaHCO3, пропустили через целит, сушили над MgSO4 и упаривали в вакууме. Полученные кристаллы очистили на столбике силикагеля (элюент ПЭ-ЭА 10:1). Получили 2.55 г (85%) соединения (4а) в виде серых кристаллов. Т.пл. 54-55 oC (ПЭ-ЭА) (см. лит. [25]: 55–56 °С). 1H NMR (300.13 MHz, CDCl3): δ 2.61 (3H, с, S-Me). 13С NMR (150.93 MHz, CDCl3): δ 173.2, 167.4, 158.6 (кв, 2JC,F= 35 Hz, C6-CF3), 119.7 (кв, 1JC,F= 278 Hz, CF3), 83.7, 14.6. 19F NMR (282.38 MHz, CDCl3): δ 8.75 (с, CF3). EIMS 70 eV, m/z: 355 [M + H]+ (47), 354 [M]+ (100), 334 [M – HF]+ (28). Вычислено для C6H3ClF3IN2S: C, 20.33; H, 0.85; N, 7.90. Найдено C, 20.39; H, 0.78; N, 7.98.
6-Дифторметил-5-иод-2-метилтио-4-хлорпиримидин (4b).
Соединение (4b) получено аналогично исходя из соединения (3b) (1.91 г, 6 ммоль), хлорокиси фосфора (20 мл) и ДБУ (0.46 г, 3 ммоль) в виде серых кристаллов. Выход 92%. Т.пл. 74-76 oC (ПЭ-ЭА). 1H NMR (300.13 MHz, CDCl3): δ 6.70 (1H, t, J = 54 Hz, CF2H), 2.61 (3H, s, S-Me). 13С NMR (150.93 MHz, CDCl3): δ 173.5, 166.1, 161.6 (t, 2JC,F= 24 Hz, C6-CF2H), 113.7 (t, 1JC,F= 245 Hz, CF3), 85.4, 14.6. 19F NMR (282.38 MHz, CDCl3): δ -41.60 (s, CF3). EIMS 70 eV, m/z: 337 [M + H]+ (50), 336 [M]+ (100), 316 [M – HF]+ (45). Вычислено для C6H4ClF2IN2S: C, 21.41; H, 1.20; N, 8.32. Найдено C, 21.58; H, 1.18; N, 8.36.
5-Иод-2-метилтио-6-трифторметил-N-(4-фторфенил)пиримидин-4-амин (5а).
Cоединение (4a) (1.06 г, 3 ммоль) растворили в диоксане (20 мл). К раствору добавили 4-фторанилин (0.35 г, 3.15 ммоль), N,N-диизопропилэтиламин (0.4 г, 3.15 ммоль) и LiBr (0.52 г, 6 ммоль). Реакционную смесь кипятили 8 часов (контроль реакции методом ТСХ, элюент ПЭ-ЭА 4:1), охладили и упарили в вакууме. Остаток растворили в хлористом метилене (40 мл), последовательно промыли 5% HCl и насыщенным раствором NaCl. Органический слой отделили, высушили над MgSO4 и упарили в вакууме. Остаток очистили колоночной хроматографией на силикагеле (элюент ПЭ-ЭА 4:1). Получено 1.15 г (90%) соединения (5а) в виде сероватых кристаллов. Т.пл. 97-99 oC (ПЭ-ЭА). 1H NMR (300.13 MHz, CDCl3): δ 7.63 (1H, уш.с, NH), 7.55 (2H, м, Ar), 7.15 (2H, м, Ar), 2.52 (3H, s, S-Me). 19F NMR (282.38 MHz, CDCl3): δ 10.43 (3F, с, CF3), -38.64 (1F, с, F-Ar). EIMS 70 eV, m/z: 430 [M + H]+ (78), 429 [M]+ (100), 428 [M – H]+ (22), 383 [M +H, – SMe]+ (12), 382 [M – SMe]+ (18), 302 [M – I]+ (19). Вычислено для C12H8F4IN3S: C, 33.58; H, 1.88; N, 9.79. Найдено C, 33.32; H, 1.76; N, 9.80.
Аналогично из соединения (4а) и N-метиланилина получен 5-иод-N-метил-2-метилтио-6-трифторметил-N-фенил-пиримидин-4-амин (5b). Время реакции 12 ч. Выход 78%. Сероватые кристаллы. Т.пл. 130-132 oC (ПЭ-ЭА). 1H NMR (300.13 MHz, CDCl3): δ 7.40 (2H, м, Ar), 7.25 (1H, м, Ar), 7.07 (2H, м, Ar), 3.57 (3H, с, N-Me), 2.61 (3H, с, S-Me). 19F NMR (282.38 MHz, CDCl3): δ 10.61 (с, CF3). EIMS 70 eV, m/z: 425 [M]+ (13), 299 [M+H – I]+ (15), 298 [M – I]+ (100), 278 [M – I– HF]+ (12). Вычислено для C13H11F3IN3S: C, 36.72; H, 2.61; N, 9.88. Найдено C, 36.44; H, 2.20; N, 9.66.
Аналогично из соединения (4b) и N-метиланилина получен 6-дифторметил-5-иод- N-метил-2-метилтио-N-фенил-пиримидин-4-амин (5c). Время реакции 12 ч. Выход 82%. Сероватые кристаллы. Т.пл. 144-145 oC (ПЭ-ЭА). 1H NMR (300.13 MHz, CDCl3): δ 7.40 (2H, м, Ar), 7.25 (1H, м, Ar), 7.09 (2H, м, Ar), 6.69 (1H, т, J = 54.5Hz, CF2H), 3.56 (3H, с, N-Me), 2.61 (3H, с, S-Me). 13С NMR (150.93 MHz, CDCl3): δ 171.5, 163.7, 160.2 (т, 2JC,F= 25Hz, C4-CF2H), 146.0, 129.7, 126.3, 126.0, 114.6 (т, 1JC,F= 243Hz, CF2H), 73.0, 42.7, 14.4. 19F NMR (282.38 MHz, CDCl3): δ -41.33 (с, CF2H). EIMS 70 eV, m/z: 408 [M + H]+ (15), 407 [M]+ (17), 281 [M+H–I]+ (19), 298 [M – I]+ (100), 260 [M – I – HF]+ (15). Вычислено для C13H12F2IN3S: C, 38.34; H 2.97; N 10.32. Найдено C, 38.03; H, 2.77; N, 10.01.
Опыты по получению 2-метилтио-4-три(ди)фторметил-9H-пиримидо[4,5-b]индолов.
В колбе с отводом для инертного газа смешали соединение (5а) (0.3 г, 0.69 ммоль), Pd(OAc)2 (16 мг, 0.07 ммоль), три(циклогексил)фосфин (40 мг, 0.14 ммоль) и ацетат натрия (115 мг, 1.4 ммоль). К смеси добавили 10 мл сухого ДМФА, полученную суспензию перемешивали 30 мин под аргоном при 20 °С, затем нагрели до 125 °С и выдержали при этой температуре 4 ч. Реакционную массу охладили до 20 °С, вылили в 250 мл холодной воды и экстрагировали этилацетатом (2 х 30 мл). Органические экстракты объединили, промыли насыщенным раствором NaCl, высушили над MgSO4 и упарили. Остаток нанесли на колонку с силикагелем. Первым элюировался продукт деиодирования - 2-метилтио-6-трифторметил-N-(4-фторфенил)-пиримидин-4-амин (6а) в виде белых кристаллов. Получено 0.13 г (60%). Белые кристаллы. Т.пл. 112-113 oC (ПЭ-ЭА). 1H NMR (300.13 MHz, CDCl3): δ 7.34 (2H, м , Ar), 7.14 (2H, м, Ar), 7.00 (1H, уш.с, NH), 6.52 (1H, s, H5), 2.57 (3H, s, S-Me). 19F NMR (282.38 MHz, CDCl3): δ 6.91 (3F, s, CF3), -37.47 (1F, м, F-Ar). EIMS 70 eV, m/z: 304 [M + H]+ (34), 303 [M]+ (100), 302 [M – H]+ (31), 257 [M +H – SMe]+ (25), 256 [M – SMe]+ (45). Вычислено для C12H9F4N3S: C, 47.52; H, 2.99; N, 13.86. Найдено C, 47.25; H, 2.66; N, 13.77.
Затем элюировался 2-(метилтио)-4-трифторметил-6-фтор-9H-пиримидо[4,5-b]индол (1а) в виде белых кристаллов. Получено 0.052 г (25%). Т.пл. >250oC. 1H NMR (300.13 MHz, d6-DMSO): δ 12.98 (1H, с, NH), 7.70 (1H, м, Ar), 7.62 (1H, дд, J= 7.6 Нz, J= 3.4 Нz, Ar), 7.50 (1H, тд, J= 8.2 Нz, J= 1.7 Нz, Ar), 2.65 (3H, c, S-Me). 19F NMR (282.38 MHz, d6-DMSO): δ 9.54 (3F, с, CF3), -44.32 (1F, с, F-Ar). EIMS 70 eV, m/z: 302 [M + H]+ (20), 301 [M]+ (100), 300 [M – H]+ (35), 268 [M – S – H]+ (17), 255 [M + H – SMe]+ (30), 186 [M + H – SMe– CF3]+ (36). Вычислено для C12H7F4N3S: C, 47.84; H, 2.34; N, 13.95. Найдено C, 47.64; H, 2.17; N, 13.84.
Аналогично был получен 9-метил-2-(метилтио)-4-трифторметил-9H-пиримидо[4,5-b]индол (1b). Белые кристаллы. Выход 48%. Т.пл. 143-145oC. 1H NMR (300.13 MHz, CDCl3): δ 8.26 (1H, д, J= 7.8 Нz, Ar), 7.68 (1H, т, J= 7.6 Нz, Ar), 7.54 (1H, д, J= 8.2 Нz, Ar), 7.46 (1H, т, J= 7.8 Нz, Ar), 4.00 (3H, с, N-Me), 2.79 (3H, с, S-Me). 13С NMR (150.93 MHz, CDCl3): δ 168.1, 157.4, 146.2 (кв, 2JC,F= 37.6Hz, C4-CF3), 140.4, 128.4, 123.4 (кв, 5JC,F= 8.9Hz), 122.4, 121.5 (кв, 1JC,F= 275Hz, CF3), 117.3, 109.6, 107.0, 28.0, 14.5. 19F NMR (282.38 MHz, CDCl3): δ 9.84 (с, CF3). EIMS 70 eV, m/z: 298 [M + H]+ (40), 297 [M]+ (100), 296 [M – H]+ (27), 264 [M – HS]+ (14), 251 [M + H – SMe]+ (42), 182 [M – SMe– CF3]+ (49). Вычислено для C13H10F3N3S: C, 52.52; H, 3.39; N, 14.13. Найдено C, 52.49; H, 3.47; N, 13.97.
Аналогично был получен 4-дифторметил-9-метил-2-(метилтио)-9H-пиримидо[4,5-b]индол (1c). Белые кристаллы. Выход 55%. Т.пл. 140-141 oC. 1H NMR (300.13 MHz, CDCl3): δ 8.26 (1H, д, J= 7.8 Нz, Ar), 7.54 (1H, т, J= 7.6Нz, Ar), 7.39 (1H, д, J= 8.2Нz, Ar), 7.36 (1H, т, J= 7.8 Нz, Ar), 6.80 (1H, т, J = 54.5Hz, CF2H), 3.85 (3H, с, N-Me), 2.71 (3H, с, S-Me). 13С NMR (150.93 MHz, CDCl3): δ 167.6, 157.1, 151.2 (т, 2JC,F= 30Hz, C4-CF2H), 140.1, 127.8, 124.4 (т, 5JC,F= 5.5Hz), 122.1, 117.6, 115.6 (т, 1JC,F= 241Hz, CF2H), 109.4, 106.9, 27.8, 14.4. 19F NMR (282.38 MHz, CDCl3): δ -40.0 (с, CF2H). EIMS 70 eV, m/z: 280 [M + H]+ (24), 279 [M]+ (100), 278 [M – H]+ (35), 246 [M – HS]+ (20), 233 [M + H – SMe]+ (42), 213 [M – SMe– HF]+ (13), 182 [M – SMe– CF2]+ (49). Вычислено для C13H11F2N3S: C, 55.90; H 3.97; N 15.04. Найдено C, 55.45; H, 3.88; N, 14.72.
Рентгеноструктурный эксперимент.
Монокристаллы соединения (1с), пригодные для рентгеноструктурного анализа, были получены медленным испарением раствора (1с) в хлороформе при 20 °С. Кристаллы триклинные, кристаллографические данные C13H11F2N3S - пространственная группа P-1: a = 8.4844(10) Å, b = 8.8967(10) Å, c = 9.4970(11) Å, α= 115.503(2)°, β =104.329(2)°, γ = 95.782(2)°, V = 609.02(12) Å3, Z = 2, M = 279.31, dcalc = 1.523 g * cm-3, μ = 0.279 mm-1. 7896 рефлексов были собраны на дифрактометре SMART APEX II CCD (λ(Mo-Kα)=0.71073 Å, графитовый монохроматор, ω-сканы, 2θ<54°) при 120K. Структура была решена прямым методом и уточнена полноматричным методом наименьших квадратов в анизотропном приближении. 2654 независимых рефлексов [Rint = 0.0226] были использованы для уточнения; wR2 = 0.0971 вычислено по F2hkl (GOF = 1.051, R1 = 0.0368 вычислено по Fhkl для 2304 отражений с I>2σ(I)). Координаты атомов, длины и углы связей, тепловые параметры депонированы в Cambridge Crystallographic Data Centre (CCDC), reference number 1031474. Эти данные доступны бесплатно через ресурс www.ccdc.cam.ac.uk/data_request/cif.
Список литературы
- Р. Г. Глушков, В. А. Вольскова, О. Ю. Магидсон, Хим-фарм. Журн. 1967, 9, 25-32 (Pharm. J. Chem. 1967, 517-523).
- G.L. Bundy, L.S. Banitt, P.J. Dobrowolski, J.R. Palmer, T.M. Schwartz, D.C. Zimmermann, M.F. Lipton, M.A Mauragis., M. F. Veley, R. B. Appell, R. C. Clouse, E. D. Daugs, Org. Process Res. Dev., 2001, 5, 144-151.
- C.E. Mueller, U. Geis, B. Grahner, W. Lanzner, K. Eger, J. Med. Chem., 1996, 39, 2482-2491.
- H. D. H. Showalter, A. J. Bridges, H. Zhou, A. D. Sercel, A. McMichael, D. W. Fry, J. Med. Chem, 1999, 42, 5464-5474.
- P.M. Traxler, P. Furet, H. Mett, E. Buchdunger, T. Meyer, N. Lydon, J. Med. Chem., 1996, 39, 2285-2292.
- N. Zaware, H. Sharma, J. Yang, R. Kumar Vyas Devambatla, S. F. Queener, K.S. Anderson, A. Gangjee, ACS Med. Chem. Lett., 2013, 4, 12, 1148–1151
- A. Gangjee, N. Zaware, R. K. V. Devambatla, S. Raghavan, C. D. Westbrook, N. F. Dybdal-Hargreaves, E. Hamel, S. L. Mooberry, Bioorg. Med. Chem. 2013, 21, 891–902.
- О.Н. Зефирова, А.Г. Дийков, Н.В. Зык, Н.С. Зефиров, Известия AН. Сер. хим. 2007, 655–662 (Russ. Chem. Bull., Int. Ed. 2007, 56, 4, 680-688), (b) Y. Lu, J. Chen, M. Xiao, W. Li, D. D. Miller, Pharm Res. 2012, 29, 11, 2943–2971.
- А. Hoffmann-Roeder, E. Schweizer, J. Egger, P. Seiler, U. Obst-Sander, B. Wagner, M. Kansy, D. W. Banner, F. Diederich, ChemMedChem., 2006, 1, 1205 – 1215.
- O. Iaroshenko, Synthesis 2009, 23, 3967–3974.
- G. Xu, L. Zheng, S. Wang, Q. Dang, X. Bai, Synlett, 2009, 3206-3210.
- C. L. Gibson, J. K. Huggan, A. Kennedy, L. Kiefer, J. H. Lee, C.J. Suckling, C. Clements, A. L. Harvey, W. N. Hunter, L.B. Tulloch, Org. Biomol. Chem. 2009, 7, 1829–1842.
- (а) A. S. Kumar, P. V. Amulya Rao, R. Nagarajan, Org. Biomol. Chem., 2012, 10, 5084-5093, (b) В. П. Боровик, O. П. Шкурко, Известия AН. Сер. хим. 2002, 1974–1977 (Russ. Chem. Bull., Int. Ed. 2002, 51, 11, 2129-2133), (c) синтез of 9H-пиримидо[4,5-b]индолов из пиримидинов, см. литературу, цитированную в M. Adib, B. Mohammadi, H. R. Bijanzadeh, Synlett, 2008, 177-180
- B. Dotzauer, R. Gruenert, P. J. Bednarski, H. Lanig, J. Landwehra, R. Troschuetz, Bioorg. Med. Chem., 2006, 14, 7282–7292.
- (а) C.E. Wright, 9H-Pyrimido[4,5-b]indole-2,4-diones, J. Heterocycl. Chem., 1976, 13, 539-544; (b) C.E. Wright, J. Gambino, J. Heterocycl. Chem., 1979, 16, 401-402.
- M. A. Mauragis, M. F. Veley, M. F. Lipton, Org. Process Res. Dev., 1997, 1, 39-44.
- I. P. Beletskaya, A. V. Cheprakov, Chem. Rev., 2000, 100, 3009-3066.
- Y.-M. Zhang, T. Razler., P. F. Jackson, Tetrahedron Lett., 2002, 43, 8235-8239.
- N. Ple, A. Turck, A. Heynderickx, G. Queguiner, J. Heterocycl. Chem., 1994, 31, 1311-1315.
- Пат. JP1495267, 1964 (Chem. Abstr., 1968 , vol. 68, # 105224h).
- T. Sakamoto, Y. Kondo, R. Watanabe, H. Yamanaka, Chem. Pharm. Bull., 1986, 34, 7, 2719-2724.
- Пат. WO2010/22121 A1 2010, A. Arasappan, G.F. Njoroge, F. Bennett, V. M. Girijavallabhan, Y. Huang, R. Huelgas, J. J. Piwinski, N.-Y. Shin, V. Verma, F. Velazquez, S. Venkatraman, C. D. Kwong, S. Ananthan, J. Clark, F. Geng, H. S. Kezar, III., J. A. Maddry, R. C. Reynolds, A. Roychowdhury, J. A. Secrist, III., A.T. Fowler.
- H. Gershon, A. T. Grefig, A.A. Scala, J. Heterocycl. Chem., 1983, 20, 219-223.
- J. J. Li, J. Org. Chem. 1999, 64, 8425-8427.
- Пат. US2005/38041 A1, 2005, Y. Nakagawa, S. Bobrov, C. R. Semer IV, T. A. Kucharek, M. Hamamoto
Статья рекомендована к публикации членом редколлегии проф. Н.Д. Чканиковым
Fluorine Notes, 2014, 97, 3-4


