Поступило в редакцию: октябрь 2012
Fluorine Notes, 2014, 95, 1-2
Химия и технология полифторированных органических соединений на основе новых агрессивостойких катализаторов
В.Ю. Захаров
Федеральное государственное бюджетное образовательное учреждение высшего профессионального образования«Вятский
государственный университет» (ФГБОУ ВПО «ВятГУ»), 610000, г. Киров, ул. Московская 36
e-mail:
zakhar.05@mail.ru
Аннотация. Разработаны научные принципы подбора эффективных катализаторов прямого газофазного фторирования органических соединений и экспериментально показана возможность резкого увеличения селективности и скорости фторирования неразбавленным фтором при направленном изменении природы каталитической композиции. Применение созданных катализаторов позволило усовершенствовать существующие и разработать новые ресурсосберегающие, экологически более чистые технологии целого ряда полифторированных органических продуктов.
Ключевые слова: Фторорганические соединения, фтор, гетерогенный катализатор, каталитическое фторирование, изомеризация.
I. Введение
Фторорганические соединения как материалы, обладающие рядом уникальных свойств, находят широкое применение в различных областях техники. Фторопласты, фторкаучуки, специальные фторированные смазки и жидкости незаменимы при работе с агрессивными средами в экстремальных условиях – использование этих материалов привело, в частности, к перевороту в технологии атомной промышленности, в космической и реактивной технике, а также высокоскоростной авиации.
Особые физико-химические свойства фторхлорорганических жидкостей (т.н. хладонов или фреонов) обеспечили бурное развитие холодильной техники.
Композиции с использованием полифторированных органических соединений («легкая вода») составляют основу современных пламягасящих средств.
Водные эмульсии ряда фторорганических жидкостей, в частности, перфтордекалина, являются эффективными переносчиками кислорода; это создает перспективы для их использования в медицине в качестве кровезаменителей. Высокая биологическая активность ряда полифторированных органических соединений используется как в медицине (противораковые препараты), так и в сельском хозяйстве (пестициды). Уже этот, далеко не полный перечень областей применения фторорганических продуктов позволяет оценить их прикладное значение.
Обширная область перфторированных продуктов может быть получена фторированием полифторированных органических соединений элементарным фтором. В то же время исследования по взаимодействию фтора с органическими соединениями сдерживаются неселективным, взрывным характером этого процесса, обусловленным его высокой экзотермичностью.
Одним из путей увеличения селективности прямого фторирования является применение гетерогенных катализаторов; следует отметить, однако, что достигнутый уровень техники каталитического фторирования хотя и позволяет существенно повысить селективность по сравнению с гомогенным газофазным процессом, тем не менее, характеризуется, как правило, высокой деструкцией субстрата, а подчас и противоречивыми, невоспроизводимыми результатами. Разработанные (известные) катализаторы оказались недостаточно эффективными для их использования в укрупненном, промышленном масштабе, а метод каталитического фторирования, как следствие, не получил широкого практического распространения. Следует отметить также, что высокая реакционная способность фтора резко сужает круг материалов, пригодных для приготовления стабильных катализаторов. Все это подчеркивает актуальность исследований, направленных на создание эффективных агрессивостойких гетерогенных контактов и развития на их основе техники прямого фторирования до уровня, обеспечивающего возможность ее широкомасштабного практического использования. Решение этой важной задачи сдерживалось, на наш взгляд, отсутствием новых рациональных подходов к подбору катализаторов прямого газофазного фторирования органических соединений.
Селективность и скорость целого класса практически важных термических превращений полифторированных органических соединений (окисление, дехлорирование водородом, изомеризация и т.д.) также могут быть увеличены при использовании эффективных катализаторов, однако, практическое применение катализа для решения актуальных задач технологии фторпродуктов по снижению материало- и энергоемкости, уменьшению загрязнения окружающей среды, синтезу новых уникальных материалов явно недостаточно. Одной из основных проблем при создании стабильных, промышленных катализаторов термических превращений полифторированных органических соединений также является повышенная агрессивность реакционной среды, что приводит к преждевременному разрушению гетерогенных контактов. Все это обуславливает актуальность исследований в области подбора эффективных агрессивостойких катализаторов термических превращений полифторированных органических соединений.
В первой части настоящей работы разработаны научные принципы подбора эффективных катализаторов прямого газофазного фторирования органических соединений и впервые экспериментально показана возможность резкого увеличения селективности и скорости фторирования неразбавленным фтором при направленном изменении природы каталитической композиции. Подобранная универсальная агрессивостойкая каталитическая композиция NiF2/α-Al2O3 позволила не только осуществить с количественными выходом синтез целого ряда практически важных перфторуглеродных продуктов (2-перфторметилперфтропентана, 3-перфторизопропил-2-перфторметилперфторпентана, перфторэтана, перфторбутана, пентафторхлорэтана и т.д.), но и впервые широко внедрить в промышленную практику технологию прямого каталитического фторирования (производство перфтордиметилперфторциклогексана, очистка перфтордекалина, жидкостей Б-I, М-I, смазки УПИ, регенерация фторуглеродных жидкостей и т.д.).
Во второй части работы на основе систематического исследования широкого класса термических газофазных превращений полифторированных органических соединений установлено, что α-AI2O3 является универсальным агрессивостойким носителем для катализаторов этих процессов. На основе α-AI2O3 разработан ряд новых эффективных гетерогенных контактов, в частности, для катализа дехлорирования водородом 1,2-дихлоргексафторпропана и 1,2-дифтортетрахлорэтана, а также окисления тетрафторэтилена.
В результате исследования кинетических особенностей термических газофазных реакций полифторированных органических соединений установлено также, что дегидратированный активированный уголь является эффективным агрессивостойким катализатором нуклеофильного типа и может быть широко использован даже в немодифицированном виде, в частности, для катализа изомеризации гексафторпропиленоксида, деструкции олигомеров карбонилдифторида, перфторированных полипероксидов и ω-хлорперфторалкилфторсульфатов, гидролиза и гирдофторирования перфторизобутилена.
Применение созданных термоагрессивостойких катализаторов позволило усовершенствовать существующие и разработать новые ресурсосберегающие, экологически более чистые технологии целого ряда полифторированных органических продуктов (гексафторпропилена, 1,2-дифтордихлорэтилена, карбонилдифторида, перфторполиоксаметиленацетилфторидов и т.д.).
2. Прямое каталитическое фторирование полифторированных органических соединений
2.1. Аналитический обзор
Химизм прямого фторирования органических соединений, которое в соответствии с /1-3/ протекает по радикальному типу, во многом определяется термодинамическими особенностями его отдельных стадий:
F2 → 2F• +155 кДж/моль
RH+F• → R•+HF – 140 кДж/моль
R• + F2 → RF+F• – 285 кДж/моль
Эти особенности заключаются в относительно низкой энергии диссоциации молекулярного фтора (155 кДж/моль) и высокой теплоте, выделяющейся при фторировании (более 420 кДж/моль фтора). Если принять во внимание, что энергия связей С-С в молекулах углеводородов составляет примерно 330 кДж-моль, становится ясным, почему для прямого фторирования весьма характерно разветвление цепей с деструкцией углеродного скелета субстрата.
Для увеличения селективности прямого фторирования применяют разбавление фтора инертными газами /4-11/, растворение органического субстрата в инертном по отношению к фтору растворителе /12-18/, охлаждение реакционных смесей /19-32/, использование реакторов специальных конструкций /33-37/ – струйного, с зонным обогревом, а также контролируемое фторирование в твердой фазе /38-51/.
Параллельно прямому фторированию развивались электрохимический /52-58/ и металлофторидный методы /59-70/; последний основан на использовании в качестве фторирующих агентов фторидов металлов, главным образом кобальта, в состоянии их высшей валентности. Использование этих, более мягких по сравнению с элементарным фтором фторирующих агентов позволяет надежно контролировать температуру фторирования, что особенно важно при осуществлении процесса в укрупненном масштабе. Именно поэтому металлофторидный метод, несмотря на повышенную материало- и энергоемкость, а также низкую производительность, лег в основу существующей технологии фторирования органических соединений.
Одним из путей увеличения селективности фторирования является осуществление взаимодействия элементарного фтора и органического субстрата в слое катализатора /71/. Во время второй мировой войны этот метод лег в основу первого производства стратегически важных фторуглеродов /72/.
Основная функция катализатора (насадки), как следует из /73-75/, заключается в рассеивании тепла, выделяющегося при фторировании; это позволяет избежать локальных перегревов, понизить среднемассовую температуру реакционных газов и, тем самым, уменьшить вклад деструкции исходного сырья, промежуточных и конечных продуктов. В этой связи становится понятным широкое использование металлических катализаторов – меди, серебра, никеля, обладающих повышенной теплопроводностью /2,72-79/.
Впервые использование металлического катализатора при фторировании, в соответствии с /2/, описано Фреденхагеном и Каденбахом: фтор в смеси с азотом (мольное отношение 1:1) вводили в стеклянный реактор (диаметр – 6см, длина – 40см) по перфорированной медной трубке, обернутой несколькими слоями медной сетки. Реактор вращали с такой скоростью, чтобы помещенная на дно органическая жидкость распределялась в виде пленки по его стенкам. Впоследствии Бигелоу использовал реактор аналогичной конструкции (диаметр – 2,8см, длина – 75см, ввод фтора и азота в мольном соотношении 1:1,6 по перфорированной медной трубке, обернутой медной сеткой) для фторирования тетрахлорэтилена и гексахлорэтана. Субстрат помещали на дно реактора и также закрывали медной сеткой; выход 1,2-дифтортетрахлорэтана – продукта селективного присоединения фтора по двойной связи тетрахлорэтилена, достигал при этом 20%.
В /2,73,80/ приведены данные по газофазному прямому фторированию этана, метана, хлористого этила, 1,2-дихлорпропана, бензола, толуола, бензотрифторида и ацетона в слое плотной насадки из роликов медной сетки или медной проволоки. Фторированием этана, кроме основного продукта – тетрафторметана, были получены гекса-, пента-, тетра- и трифторэтан; образования ди- и монофторэтана не наблюдалось. При фторировании метана, кроме тетра-, три-, ди- и монофторметана, было зафиксировано образование гексафторэтана и октафторпропана, что свидетельствует об ассоциации промежуточных радикалов /2,73/.
При фторировании хлористого этила в продуктах реакции, кроме тетрафторметана и продуктов селективного замещения атомов водорода на фтор, были обнаружены 1,1-дихлорэтан и 1,1-дихлор-2,2-дифторэтан. Этот результат указывает на то, что во время фторирования имеет место и хлорирование; последнее происходит или благодаря образованию хлористого фтора, являющегося сильным хлорирующим агентом, или вследствие взаимодействия субстрата с выделяющимся свободным хлором /73/.
Аналогичные результаты были получены при газофазном фторировании трихлорэтилена над измельченной медью /2/ - одним из основных продуктов реакции является тетрахлордифторэтан. В соответствии с /80/ одними из основных продуктов прямого фторирования 1,2-дихлорпропана в слое насадки из медной проволоки при 420-470 К являются трихлорпентафторпропан и тетрахлортетрафторпропан; выход 1,2-дихлоргексафторпропана – продукта селективного замещения атомов водорода на фтор, составляет в этих условиях 15-25%.
Для прямого газофазного фторирования ароматических соединений в слое медной насадки весьма характерны деструкция углеродного скелета и полимеризация; так, из продуктов фторирования бензола были выделены CF4, C2F6, C3F8, C4F10, C5F12, C6F12, C6НF11 и C12F22 /73/. Выход перфтордекалина при фторировании тетралина (620-650 К) над посеребренной медной стружкой составил всего 15% /81/. При фторировании α-метилнафталина над посеребренной медной проволокой образуются лишь незначительные количества перфторметилперфтордекалина /73/. При фторировании бензотрифторида в аналогичных условиях кроме перфторметилперфторциклогексана образуются значительные количества продуктов деструкции и неполного фторирования, в том числе перфторциклогексан и перфторметилмоногидроперфторциклогексан /73/.
В то же время в /72/ отмечается, что модифицирование медных стружек серебром (реже – золотом) позволяет достичь существенного увеличения выхода продуктов селективного исчерпывающего фторирования (Табл. 1).
Прямому фторированию этим методом были подвергнуты также 2,2,3-триметилбутан, 2,2,4-триметилпентан, цетан, толуол, мезитилен, ретен и хризен /72/. Характеристики продуктов их селективного фторирования приведены в /82/.
В /73/ сформулированы следующие условия для достижения максимального выхода фторуглеродов при прямом фторировании:
- применение катализатора – серебра на металлической насадке;
- осуществление процесса в избытке фтора;
- поддержание температуры в пределах от 410-600 К (при температуре ниже 410 К органические вещества конденсируются на насадке и деструктируют во фторе, а при температуре выше 600 К разрушается насадка);
- фтор и пары органического субстрата должны смешиваться в слое катализатора.
Таблица 1. Фторирование углеводородов над посеребренной медной насадкой /72/.
|
Исходное вещество |
Температура реакции, К |
Выход продукта селективного исчерпывающего фторирования, % от теоретического |
|
C6H6 |
538 |
58 |
|
н-C7H16 |
408 |
62 |
|
C6H5CF3 |
473 |
85 |
|
C6H4(CF3)2 |
473 |
87 |
|
Антрацен |
573 |
43 |
|
Нафтеновое масло |
563 |
15 |
|
Ароматическое масло |
573 |
19 |
В соответствии с /2/, фторид меди, покрывающий медную насадку, не является, как показали прямые эксперименты, фторирующим агентом – это, по мнению авторов /2/, сводит функцию медной насадки к устранению локальных перегревов и исключает возможность ее каталитического действия. Дифторид серебра, напротив, является эффективным фторирующим агентом /2,61/. В этой связи авторы /72/ полагают, что основным фторирующим агентом при использовании модифицированной серебром медной насадки является фторное серебро, а роль элементарного фтора сводится к регенерации этого фторида.
В то же время авторы /74,75/ приходят к противоположным выводам и полагают, что модифицирование медной насадки серебром не оказывает сколько-нибудь существенного влияния на ее каталитические свойства. Сравнительные данные по фторированию бензола при 538 К на различных металлических катализаторах /74/ приведены ниже (табл. 2).
Из приведенных данных видно, что каталитическое действие большинства металлических насадок примерно одинаково; видно также, что серебрение практически не сказывается на каталитических свойствах медных стружек.
Таблица 2. Прямое каталитическое фторирование бензола.
|
Катализатор |
Выход C6F12, % от теоретического |
|
Медные стружки |
29 |
|
Посеребренные медные стружки |
26 |
|
Позолоченные медные стружки |
35 |
|
Никелированные медные стружки |
23 |
|
Медные стружки, покрытые кобальтом |
28 |
|
Латунные стружки |
24 |
|
Амальгамированные медные стружки |
10 |
|
Медная проволока, покрытая родием |
17 |
|
Хромированная медная проволока |
7 |
В /2/ отмечается, что в отдельных случаях модифицированные катализаторы оказались даже менее эффективным, чем непокрытая медная и никелевая дробь, и делается вывод о том, что главная функция металлической насадки состоит в рассеивании тепла реакции, вследствие чего фторирование протекает при умеренных температурах. Отмечается также /2/, что форма насадки (сетка, стружка, дробь) является не менее важным фактором, чем природа металлической поверхности.
В качестве катализатора прямого фторирования ароматических соединений описан также трифторид кобальта /83-85/. Обработка α-метилнафталина элементарным фтором при 633 К в слое трифторида кобальта сопровождается в основном деструкцией; выход перфторметилперфтордекалина при этом составил всего 11%. Фторирование перхлорциклопентадиена в тех же условиях приводит к образованию пентахлорпентафторциклопентана с выходом 70% /83/.
Сообщается о получении жидких фторуглеродов путем исчерпывающего фторирования пентана, гексана, толуола, мета-ксилола, тетралина, α-метилнафталина /84,85/, алкиладамантанов, адамантанкарбоновых кислот, производных бициклогептана, бициклононана /86/; субстрат испаряют и пропускают через псевдоожиженный слой трифторида кобальта при 600-725 К в присутствии элементарного фтора. Стадии фторирования элементарным фтором может предшествовать частичное фторирование углеводородов фторидами серебра, марганца, серы или сурьмы /86/.
Электрохимическое фторирование циклоалканов или их частично замещенных фторпроизводных в безводном фтористом водороде в присутствии фторидов кобальта, никеля или марганца описано в /56/.
В /87,88/ описано фторирование органических соединений на частицах фторида щелочного металла, диспергированных в потоке инертного газа. Авторы /87/ смешивали органический субстрат с фторидом натрия, тщательно растирали, распыляли в струе азота и подавали на фторирование; в качестве субстрата использовались ароматические соединения (бензол, толуол, нафталин), алифатические и циклоалифатические углеводороды (например, гексан, додекан), полиэтилен, замещенные углеводороды (нитробензол, бензойная кислота), а также некоторые двухосновные кислоты. В /87/ отмечается существенный вклад ассоциации промежуточных радикалов при низкотемпературном фторировании ароматических соединений на фториде натрия. Так, фторированием бензола фтором, разбавленным азотом (в соотношении 1:10), при 253 К получена смесь веществ со средней молекулярной массой 670 у.е., а из толуола в аналогичных условиях – продукты со средней молекулярное массой около 1000 у.е.
Авторы /88/ предварительно готовили аэрозоль фторида натрия в струе гелия, смешивали с потоком гелия, насыщенным парами субстрата – пентана, циклогексана или диоксана, и направляли в охлажденный реактор фторирования. Фторированием циклогексана, например, при 210 К, в соотношении фтор:субстрат, равном 1:1 и десятикратном разбавлении фтора гелием получены монофторциклогексан с выходом 26%, дифторциклогексан (20%), а также другие полифторированные продукты. Для регулирования селективности фторирования авторы /88/ применяли реактор с многосекционным охлаждением, позволяющим в каждой зоне реакционного пространства поддерживать определенную температуру. Прямое фторирование азот-, кислород- и серусодержащих органических соединений на фторидах щелочных металлов описано также в /89-97/.
В /98-100/ описано каталитическое фторирование 2-гидроперфторпропана на меди, серебре, золоте, ртути, кобальте, платине, их фторидах или сплавах с получением октафторпропана. Первоначально исходное сырье – гексафторпропилен обрабатывают фтористым водородом при 370-570 К в присутствии оксифторида хрома. Образующийся при этом 2-гидроперфторпропан контактируют при 370-620 К в слое катализатора с элементарным фтором (избыток фтора 5-30%), разбавленным азотом (1:1). Выход октафторпропана при 510-520 К на покрытых серебром медных стружках, предварительно обработанных фтором, составил почти 95% от теоретического (на прореагировавший гексафторпропилен).
Получение октафторпропана прямым фторированием пропана или пропилена при 330-510 К в присутствии плавленого пористого оксида алюминия описано в /101, 102/. Фтор вводят во внутреннюю часть трубы, изготовленной из плавленого оксида алюминия, через поры которой он мигрирует во внешнюю часть, куда подают углеводород. Выход октафторпропана при этом составляет 10-85% от теоретически возможного /101/. В качестве пористого элемента описаны также металлические медь и никель /102, 103/; пористая труба, через которую мигрирует фтор, может быть пропитана каталитически активными веществами /104/.
В качестве катализаторов селективного замещения атомов галоида (в основном, хлора) на фтор при прямом фторировании органических соединений используют фториды кобальта /73/, алюминия /105/, сурьмы и титана /106/, а также хлорное железа /107/. Так, авторы /105/ использовали в качестве катализатора трифторид алюминия (в виде кристаллов размером 500Ǻ), который предварительно контактировали с кислородом при 670-730 К /108/; фторирование осуществляли в температурном интервале от 380 К (на входе в реактор) до 630 К (на выходе из реактора) при мольном соотношении хлорфторуглерод: фтор, равном 1:1,7, и времени контактирования 125 сек. Применение фторида алюминия позволило из 1,2,2-трихлорперфторпропана получить перфторпропан с выходом 81%, а из 2,3,3-трихлорперфторбутана – перфторбутан (выход 76%).
Первым успешным опытом по прямому газофазному фторированию кислородсодержащих органических соединений считают /73/ – работу Бигелоу и сотр., где в качестве субстрата использовали ацетон, а в качестве катализатора – медную сетку. Кроме перфторацетона и продуктов неполного фторирования ацетона, реакционные газы содержали значительные количества продуктов деструкции – тетрафторметана, карбонилдифторида и перфторацетилфторида. При соотношении фтор:ацетон:азот, равном 6:1:19 и начальной температуре в реакторе 333 К, выход перфторацетона составил 10%. Фторирование ацетона более концентрированным фтором (фтор:азот=1:1) при температуре 473 К в реакторе, заполненном позолоченными или платинированными медными стружками, характеризуется увеличением выхода продуктов деструкции. Фторированием метилэтилкетона в аналогичных условиях с выходом около 10 % получен октафторбутанон-2 /2/.
Прямое фторирование спиртов, в соответствии с /73/, впервые осуществлено Кэди и сотр.; основными продуктами фторирования метилового спирта разбавленным фтором при 450 К в присутствии посеребренной медной ленты были карбонилдифторид и трифторметилгипофторит. В /109-120/ приведены данные по получению трифторметилгипофлорита, дифторметилен-бис-гипофторита и перфторалкилгипофторитов селективным присоединением фтора по двойной связи углерод-кислород; эффективными катализаторами этого процесса являются фториды щелочных металлов (особенно – цезия), активные даже при 195 К и ниже, а также фториды серебра и никеля.
В /121, 122/ приведены результаты по прямому фторированию азотсодержащих органических соединений. Основным продуктом фторирования метиламина при 370 К (соотношение фтор:амин:азот равнялось 5:1:15) в слое медной дроби был тетрафторметан; при этом образуются также CF3NF2, CF3CF2NF2 и (CF3)2NF /121/. Состав продуктов реакции свидетельствует о значительной деструкции сырья и последующей частичной ассоциации образующихся радикалов.
Фторирование малононитрила при 523 К в слое медной дроби (соотношение фтор:малононитрил:азот равнялось 3:1:9) характеризуется образованием тетрафторметана (13 масс. %), октафторпропана (30 масс. %), а также продуктов деструктивного фторирования и ассоциации образующихся радикалов /122/.
Прямому каталитическому фторированию был подвергнут ряд сераорганических соединений ; так, в /71/ сообщается, что взаимодействие метилмеркаптана с элементарным фтором при 437 К в слое модифицированной серебром медной сетки сопровождается образованием (с невысоким выходом) продуктов селективного замещения атомов водорода – CF3SF5 (15%) и CF3SHF4 (15%).
Таким образом, прямое газофазное фторирование органических соединений характеризуется, как правило, значительным вкладом деструктивного фторирования с разрывом С-С связей в молекуле субстрата, что обусловлено высокой энергией, выделяющейся в ходе элементарных стадий этого экзотермического процесса. Для уменьшения деструкции при газофазном фторировании применяют гетерогенные катализаторы – в основном, металлы (как правило, медь), которые в ряде случаев модифицируют (чаще всего – серебром). Выбор металлических катализаторов обусловлен их повышенной теплопроводностью, что способствует эффективному отводу тепла из зон локальных перегревов и, соответственно, уменьшению деструкции.
В то же время следует отметить, что каталитическое фторирование хотя и позволяет существенно повысить селективность процесса (по сравнению с гомогенным газофазным фторированием), тем не менее, характеризуется, как правило, относительно высокой деструкцией сырья, а подчас и противоречивыми, невоспроизводимыми результатами. Разработанные катализаторы оказались недостаточно эффективными для их использования в укрупненном промышленном масштабе, а метод каталитического фторирования, как следствие, не получил широкого практического распространения. Это подчеркивает актуальность исследований, направленных на создание эффективных катализаторов и развитие на их основе технологии прямого фторирования до уровня, обеспечивающего возможность ее широкомасштабного практического использования. Решение этой важной задачи сдерживалось, на наш взгляд, отсутствием новых, рациональных подходов к созданию эффективных катализаторов прямого газофазного фторирования органических соединений.
2.2. Научные основы подбора эффективных агрессивостойких катализаторов прямого газофазного фторирования органических соединений
В основу развиваемого нами подхода к подбору эффективных катализаторов фторирования органических соединений положены следующие соображения:
1. Газофазное взаимодействие органических соединений с фтором приводит к образованию промежуточных аддуктов, обладающих избыточной энергией. Эти возбужденные аддукты подвергаются либо самопроизвольной деструкции, либо переходу в основное состояние за счет столкновений с молекулами газовой фазы или поверхностью.
2. Можно полагать, что улучшение селективности взаимодействия органических соединений и фтора будет достигнуто при локализации элементарных актов фторирования на поверхности. В этом случае резко увеличивается вероятность своевременного отвода избыточной энергии возбужденных аддуктов по связям с элементами поверхности насадки, которая играет роль усредняющего «теплового резервуара». При этом уменьшается деструкция возбужденных аддуктов, но не термодеструкция (например, из-за сильного разогрева реакционных газов).
3. Для локализации элементарных актов на поверхности катализатор должен не только интенсивно сорбировать молекулярный фтор, то есть обладать развитой, имеющей сродство к фтору поверхностью ( наличие развитой поверхности приведет к улучшению селективности и за счет обрыва цепей радикального разветвленного процесса), но и активировать его.
Только в этом случае возникают кинетические предпосылки для локализации процесса непосредственно на поверхности.
Таким образом, предпочтение при выборе катализатора фторирования следует отдавать контактам с развитой поверхностью, модифицированной активирующими фтор добавками. В этой связи становится понятно низкая эффективность металлических катализаторов, внешняя поверхность которых невелика.
4. Специфичной особенностью прямого фторирования является высокая агрессивность среды; это накладывает дополнительные требования к стабильности катализаторов.
Основная принципиальная особенность этого нового подхода к созданию эффективных катализаторов прямого фторирования заключается в предположении о возможности резкого увеличения селективности за счет локализации актов фторирования на поверхности, а не за счет снижения или усреднения температуры реакционных газов (снятие локальных перегревов) при использовании теплопроводной насадки – катализатора, как это делалось ранее.
Действительно, деструкция сырья часто наблюдается /19,87,88/ при фторировании в контролируемых мягких условиях (273 К и ниже), когда термический разрыв молекулярных связей маловероятен – этот эффект можно объяснить только распадом энерговозбужденных аддуктов /123,124/. Энергетическое разветвление цепей весьма характерно для низкотемпературного газофазного фторирования и определяет, в частности, кинетические особенности его протекания /125-136/. Так, существенное уменьшение скорости фторирования при переходе от дифторметана к трифторметану авторы /125-129/ объясняют возможностью разветвления цепей при фторировании CH2F2 за счет распада возбужденной молекулы образующегося CHF3:
CH2F2 + F• → CHF2 + HF
CHF2 + F2 → CHF3* + F•
CHF3* → :CF2 + HF
:CF2 + F2 → CF3• + F•
При фторировании трифторметана такая возможность отсутствует, что и обуславливает резкое уменьшение скорости процесса. В этой связи становится понятной наблюдаемая в /132-133/ высокая скорость фторирования дихлорметана и фторхлорметана – здесь также имеется возможность энергетического разветвления цепей при распаде энергонасыщенных промежуточных аддуктов.
Интересно в этой связи, что скорость фторирования дифторметана существенно снижается, а выход трифторметана увеличивается при повышении парциального давления инертного газа; этот эффект объясняется /134/ дезактивацией возбужденных молекул образующегося трифторметана при столкновении с молекулами инертного газа.
Соотношение скоростей распада энерговозбужденных при фторировании и термически активированных молекул наглядно характеризуют данные по селективности фторирования фторхлорметана /133/ – распад образующегося дифторхлорметана протекает только с отщеплением фтористого водорода, а не хлористого водорода, что характерно для термически активированной молекулы дифторхлорметана, например, при его пиролизе /137/.
Скорость распада по возбужденной связи C-F существенно выше скорости перераспределения избыточной энергии по невозбужденным связям, что и обуславливает необычную селективность деструкции образующегося дифторхлорметана /133/. Эти результаты свидетельствуют о влиянии распада образующихся возбужденных аддуктов на селективность фторирования органических соединений элементарным фтором.
Ниже приведена принципиальная кинетическая схема для обсуждаемой выше модели каталитического фторирования, анализ которой наглядно иллюстрирует требования к эффективному катализатору:
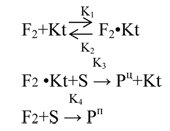
Схема имеет общий характер и не конкретизирует, например, элементарные стадии, их радикальный механизм, а описывает только основные закономерности фторирования, характеризующегося наличием конкурирующих процессов в газовой фазе и на поверхности.
При этом предполагается, что взаимодействие адсорбированного фтора (F2•Kt) с субстратом (S) на поверхности катализатора сопровождается образованием только целевых продуктов (Pц) со скоростью, характеризующейся эффективной константой К3; фторирование же с участием газообразного фтора (F2), характеризующееся константой К4, приводит к образованию только побочных продуктов (Pп).
Анализ этой схемы показывает, что величина, обратная селективности фторирования (I/α), описывается уравнением:
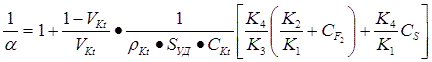
Отсюда видно, что селективному фторированию способствует рост доли реакционного объема, заполненного катализатором (VKt), увеличение его удельной поверхности (Sуд) и поверхностной концентрации активных центров (СKt), а также наличие у контакта повышенной сорбционной способности по отношению к молекулярному фтору (К2/К1) и его активирующее влияние на адсорбированный фтор (К3). Селективность, напротив, снижается при увеличении вклада газофазного фторирования (К4), а также концентраций фтора (СF2) и субстрата (Cs).
Отметим, что вытекающие из рассматриваемой модели требования к эффективному катализатору фторирования относительно универсальны; например, активация фтора может включать его диссоциацию на поверхности с образованием атомарного фтора, реакционная способность которого повышена к широкому классу (если не ко всем классам) субстратов.
Первым этапом по определению состава оптимальной каталитической композиции был подбор компонента катализатора, активирующего фтор наиболее интенсивно. В качестве модельной реакции было исследовано газофазное взаимодействие оксида углерода и фтора с образованием трифторметилгипофторита (ТГФ) – эффективного инициатора и реагента для синтеза целого ряда фторорганических соединений /138-140/.
Каталитическое фторирование оксида углерода
Газофазное фторирование оксида углерода включает две стадии:
CO + F2= COF2 –525 кДж/моль
COF2 + F2= CF3OF –135 кДж/моль
Скорость второй стадии этого процесса – взаимодействия карбонилдифторида и фтора, может быть существенно увеличена при использовании катализатора или инициатора /112-118, 141-142/.
Исследование каталитических свойств фторидов серебра, никеля, кобальта, меди, марганца, железа, цезия, калия, натрия, лития, бария, стронция, кальция и магния, нанесенных в эквимолярных количествах методом пропитки на γ-AI2O3, выявило, что активность этих контактов неодинакова и определяется природой нанесенного фторида металла. Активность коррелирует, как следует из рис. 1, с потенциалом ионизации соответствующего катиона. Этот результат можно понять, если учесть, что молекула фтора обладает ярко-выраженным электроно-акцепторным характером. Частичный переход электрона от катиона с низким потенциалом ионизации на σ-разрыхляющую орбиталь адсорбированной молекулы фтора облегчает диссоциацию галогена с образованием активного атомарного фтора /143/, что и обуславливает увеличение скорости реакции.
Наибольшей каталитической активностью, как следует из рис. 1, обладают нанесенные фториды никеля, серебра и цезия.
В /144-147/ отмечается, что никель является эффективным катализатором диссоциации молекулярного фтора на атомы. Аддукты взаимодействия никеля с фтором описываются формулой NiFx, где 2≤х≤3; их ИК-спектры совпадают со спектрами трифторида никеля /142, 145, 148/. Аддукты взаимодействия хлорида никеля с фтором также описываются формулой NiF2,5 /149/.
Полученные в /139/ кинетические данные по прямому фторированию карбонилдифторида на дифториде никеля согласуются со схемой процесса, включающей атомизацию фтора на катализаторе (порядок реакции по фтору равен 1,5). По оценкам, приведенным в /139/, скорость диссоциации фтора на дифториде никеля увеличивается более чем на пять порядков (по сравнению с некаталитической диссоциацией). Атомизация фтора на поверхности низших фторидов никеля и серебра, сопровождающая образованием высших фторидов /149/, и обуславливает, по-видимому, их каталитическую активность в прямом фторировании оксида углерода.
Каталитическое действие фторида цезия при фторировании карбонилдифторида – промежуточного продукта в синтезе ТГФ, обусловлено, как следует из /112-116/, активацией субстрата (а не фтора) с образованием соответствующего алкоксида цезия; этот результат получен при осуществлении реакции в весьма мягких условиях – 200 К и ниже.
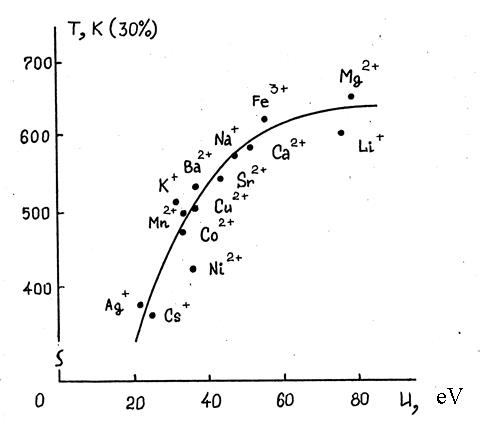
Рисунок 1. Корреляция активности нанесенных фторидов металлов в каталитическом фторировании оксида углерода и потенциалов ионизации (u, э.в.) соответствующих катионов (за меру активности принимали температуру Т, при которой концентрация трифторметилгипофторита в реакционных газах достигала 30 объемн.% при скорости подачи реагентов 1800 час-1 и мольном соотношении F2:CO=2).
В то же время в /150, 151/ отмечается, что фторалкоксиды щелочных металлов, в частности – цезия, разрушаются при температурах выше 470 К и, таким образом, в изучаемых условиях синтеза ТГФ (до 650 К) неустойчивы. Каталитическое действие фторида цезия при высокотемпературном фторировании оксида углерода до ТГФ, поэтому также может быть обусловлено активацией фтора.
Высокотемпературное каталитическое фторирование оксида углерода, в том числе и на нанесенном фториде цезия, характеризуется, как показал анализ реакционных газов, образованием заметных количеств перекиси бистрифторметила /152/, что, по мнению автора /115/, свидетельствует в пользу радикального механизма фторирования с участием атомарного фтора.
Отметим, что электрохимическое фторирование оксида углерода, карбонилдихлорида и карбонилдифторида в мягких условиях, протекающее с участием атомарного фтора, характеризуется высоким выходом ТГФ /153-154/. Этот результат указывает на повышенную активность атомов фтора при синтезе ТГФ и свидетельствует в пользу предлагаемого механизма активации фтора на активном компоненте катализатора при фторировании оксида углерода.
Характерно, что катализаторы, приготовленные механическим смешением (тщательное растирание в агатовой ступке) компонентов фторида металла и γ-AI2O3, обладают существенно меньшей активностью, чем катализаторы, полученные методом нанесения (пропитки). Этот результат обусловлен, по-видимому, различным уровнем дисперсности активного компонента и свидетельствует о существенном влиянии методики приготовления катализаторов на их эффективность. Интересно, что нанесенные катализаторы NiF2/γ-AI2O3, полученные с использованием хлорида, нитрата и сульфата никеля (при предварительном фторировании катализаторов эти соли, нанесенные на γ-AI2O3, переходят в дифторид никеля) обладают примерно одинаковой активностью.
Отметим, что известные металлические катализаторы – плотная насадка из тонкой медной проволоки и ролики из никелевой сетки, в аналогичных условиях были практически неактивны. Это является, по-видимому, следствием их низкой удельной поверхности (удельная поверхность металлических катализаторов – плотной насадки из спиралей медной проволоки и роликов из никелевой сетки составляет2,8 х 10-3 м2 и 3,5 х 10-3 м2 на 1 мл катализатора, соответственно. Для сравнения, удельная поверхность отработанных катализаторов на основеγ-AI2O3 составляет 15-20 м2/мл).
Следующим этапом определения состава оптимального катализатора прямого фторирования был выбор носителя. Рентгенофазовое исследование отработанных катализаторов на основе γ-AI2O3 и определение их химического состава показало, что в ходе реакции происходит полное фторирование контактов с образованием фазы фторида алюминия. Этот фазовый переход сопровождается уменьшением механической прочности катализаторов, что приводило к постепенному разрушению их гранул и увеличению сопротивления реактора.
Интересное наблюдение было сделано нами при изучении каталитических свойств оксида никеля, нанесенного на α-AI2O3; этот катализатор не только обладал высокой активностью при фторировании оксида углерода с получением ТГФ, но и сохранял свою первоначальную прочность и гранулометрический состав:
|
Катализатор |
Условия эксплуатации |
Суммарный вес фракции, % с размером гранул менее1, мм |
||||
|
0,5 |
2,0 |
3,0 |
5,0 |
7,0 |
||
|
NiO/γ-AI2O3 |
553 К, 24 часа |
12,6 |
45,8 |
73,2 |
91,9 |
100,0 |
|
NiO/α-AI2O3 |
623 К, 200 часов |
0,5 |
0,8 |
1,0 |
2,9 |
100,0 |
Из приведенных данных видно, что только 8,1% гранул катализатора на основе γ-AI2O3 сохранили свой размер после эксплуатации при 553 К в течение 24 часов; остальные подверглись разрушению. Катализатор на основе α-AI2O3, эксплуатируемый в более жестких условиях (623 К, 200 часов) практически не изменил свой гранулометрический состав.
Рентгенофазовый анализ катализатора NiО/α-AI2O3 выявил, что в ходе эксплуатации или предварительного фторирования нанесенный оксид полностью переходит во фторид никеля, в то время, как интенсивность линий, отвечающих носителю – α-AI2O3, практически не изменяется. Наличие в спектрах линий поглощения (малой интенсивности), отвечающих фториду алюминия, дает основание полагать, что в ходе предварительной обработки фтором и эксплуатации α-AI2O3 покрывается тонкой пленкой фторида алюминия, которая предохраняет его от дальнейшего фторирования и, тем самым, от механического разрушения. Эксплуатируемый катализатор, таким образом, представляет собой фторид никеля, нанесенный на α-AI2O3, покрытый фторидом алюминия. Для краткости в дальнейшем этот контакт обозначается как NiF2/α-Al2O3.
Отметим, что обнаруженная в работе исключительно высокая устойчивость α-AI2O3 во фторирующих средах при повышенных температурах, позволила, как будет показано ниже, использовать этот носитель для приготовления эффективных катализаторов широкого класса реакций фторорганических соединений.
Испытания промышленного катализатора марки ГИАП-3-6Н, который также представляет собой оксид никеля, нанесенный на α-AI2O, в реакции фторирования карбонилдифторида подтвердили высокую эффективность этой каталитической композиции (табл. 3) и позволили получить реакционные газы с содержанием ТГФ выше 95 объемн.%.
Таблица 3. Прямое фторирование карбонилдифторида на катализаторе ГИАП-3-6Н (объемная скорость подачи реагентов – 100 час-1; мольное отношение субстрат:фтор=1).
|
№ оп. |
Температура, К |
Состав* реакционных газов, объемн.% |
|||
|
F2+CO |
CF2O |
(CF3O)2 |
CF3OF |
||
|
1 |
448 |
2,6 |
1,7 |
0,4 |
95,3 |
|
2 |
473 |
2,2 |
1,4 |
0,2 |
96,2 |
|
3 |
498 |
1,8 |
1,1 |
<0,1 |
97,1 |
* Содержание тетрафторметана и диоксида углерода в реакционных газах ниже 0,1 объемн.%.
Отметим, что фториды серебра и цезия, нанесенные на α-AI2O3, также обладают высокой начальной каталитической активностью в синтезе ТГФ, основанном на фторировании оксида углерода; носитель α-AI2O3 и в этом случае сохраняет высокую механическую прочность даже после длительной эксплуатации в весьма жестких условиях. В то же время для этих катализаторов характерно снижение активности во времени, которое обусловлено постепенным «отслаиванием» и уносом активного компонента – фторидов металлов; последнее приводит также к увеличению сопротивления слоя катализаторов и забивкам в реакторе.
Кроме того, фториды цезия и, особенно, серебра дороги и дефицитны. В этой связи в дальнейшем основное внимание было уделено изучению каталитических свойств активного, стабильного и доступного NiF2/α-AI2O3, а также их сопоставлению со свойствами инертных насадок, например, плавленого фторида кальция, немодифицированного α-AI2O3) и известных металлических катализаторов.
Недофторированные примеси представляют собой сложную смесь полифторированных непредельных и водородсодержащих соединений. Для подбора эффективных катализаторов их прямого фторирования нами было проведено изучение модельных реакций прямого каталитического фторированя широкого класса непредельных и водородсодержащих индивидуальных полифторированных органических соединений.
Изучение модельных реакций прямого фторирования, впрочем, в ряде случаев имело самостоятельный практический интерес.
2.3. Каталитическое газофазное фторирование полифторированных соединений с двойной связью
2.3.1 Фторирование перфторноненов
Для оценки эффекта активации фтора поверхностью на скорость и селективность его присоединения по двойной связи в качестве модельной реации было изучено газофазное фторирование перфтононенов (ПФЕН).
ПФЕН образуются с высоким выходом при анионной олигомеризации гексафторпропилена (155-162):
|
|
|
3-перфторизопропил-2-перфторметилперфторпентен-2 (ПФЕН-1) 60 масс.% |
|
|
3-перфторизопропил-4-перфторметилперфторпентен-2 (ПФЕН-2) 30 масс.% |
|
|
|
2,4-перфтордиметилперфторгептен-3 (ПФЕН-3) 5 масс.% |
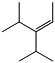
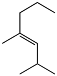
Перфторнонаны (ПФАН) – продукты селективного фторирования ПФЕН, являются перспективными диэлектриками, хладоагентами и термостойкими растворителями.
Данные по прямому газофазному фторированию ПФЕН в слое металлической меди и никеля, плавленого фторида кальция, а также α-AI2O3, модифицированного оксидом никеля, представлены в табл. 4, здесь же для сравнения приведены результаты по фторированию ПФЕН в полом реакторе без катализатора.
Особенностью прямого газофазного фторирования ПФЕН в полом реакторе, а также в слое металлической меди, никеля и плавленого фторида кальция при температурах выше 440 К является, как показатели эксперименты, его неселективный, взрывной характер; основными продуктами реакции при этом являются тетрафторметан и сажа.
Нетривиальные результаты были получены нами в ходе изучения газофазного взаимодействия ПФЕН с фтором при температурах ниже 420 К – основным продуктом реакции в полом реакторе, а также в слое металлической меди, никеля и плавленого фторида кальция при этом является (табл. 4, оп. 1-9) – стабильный радикал – 3-перфторизопропил-2-перфторметилперфторпентил-3 (ПФИЛ); он был выделен нами и охарактеризован методом ЭПР. Выход ПФИЛ при газофазном взаимодействии ПФЕН и фтора достигает 85% (на прореагировавший ПФЕН).
Этот стабильный перфторалкильный радикал впервые получен и охарактеризован методом ЭПР авторами /163,164/ при изучении жидкофазного прямого фторирования ПФЕН; жидкофазное селективное фторирование олигомеров гексафторпропилена описано также в /165,166/. Отметим, что образование стабильных перфторалкильных радикалов при радиолизе и фотолизе перфторолефинов, в частности, олигомеров гесафторпропилена, описано в /167-173/.
Характерной особенностью ПФИЛ, как показали исследования, является его чрезвычайная устойчивость – так, в частности, он стабилен при хранении на воздухе (6 месяцев). ПФИЛ устойчив даже в атмосфере фтора до 420 К (время контакта – 20 сек) как при их гомогенном смешении, так и при контактировании в слое металлических меди, никеля и плавленого фторида кальция; при увеличении температуры выше 440 К взаимодействие ПФИЛ и F2 носит взрывной характер с образованием тетрафторметана и сажи.
Контактирование ПФИЛ с фтором в слое NiF2/α-Al2O3 при 400 К приводит к образованию эквимольного количества 3-перфторизопропил-2-перфторметилперфторпентана (ПФАН), а при 520 К и выше – к образованию 3-перфторэтила-2-перфторметилперфторпентана (ПФАО) и тетрафторметана:
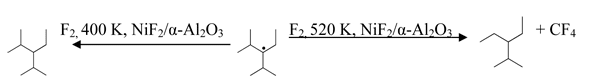
ПФИЛ полностью деструктирует в газовой фазе при 470 К за 30 сек. с образованием 3-перфторэтил-4-перфторметилперфторпентена-2 (ПФЕО) и тетрафторметана; при контактировании с фторированным α-Al2O3 в этих условиях наряду с тетрафторметаном и ПФЕО образуется ПФАО, а также ПФЕН и ПФАН:

ПФИЛ реагирует также с йодидом калия (водный раствор, 370 К, 1 час) с выделением йода и образованием ПФЕН и ПФЕО:
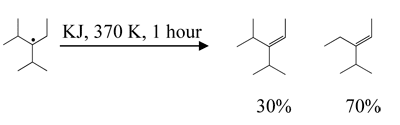
Химические свойства ПФИЛ обобщены в табл. 5.
Принципиальное изменение характера прямого газофазного фторирования ПФЕН – резкое увеличение его скорости и полное обращение селективности, было выявлено нами при испытании в качестве катализатора фторида никеля, нанесенного на α-Al2O3. Так, степень конверсии ПФЕН-1 и фтора при фторировании в полом реакторе, в слое металлических меди, никеля или плавленого фторида кальция (403 К) составляет 2-5 и 4-13%, а на катализаторе NiF2/α-Al2O3 в тех же условиях – более 99 и 88%, соответственно (табл. 4, оп. 1,3,5,7 и 12).
Фторирование в слое фторида никеля, нанесенного на α-Al2O3 при 393-493 К сопровождается количественным образованием ПФАН – продукта селективного присоединения фтора по двойной связи ПФЕН (табл. 4, оп. 10-12); на известных катализаторах – металлических меди и никеле, ПФАН в этих условиях практически не образуется.
Таблица 5. Некоторые химические свойства ПФИЛ.
|
# п/п |
Условия осуществления реакции |
Основные продукты реакции |
|
1 |
Воздух; 298 К, 6 месяцев |
Не реагирует (стабилен) |
|
2 |
Фтор, ниже 420 К, 20 сек |
Не реагирует |
|
3 |
Фтор, Cu, Ni (металл.) CaF2, <420 К |
Не реагирует |
|
4 |
Фтор, выше 440 К |
СF4, сажа |
|
5 |
Фтор, Cu, Ni (металл.) CaF2, выше 440 К |
СF4, сажа |
|
6 |
Фтор, NiF2/α-Al2O3 , 400 К, 20 сек |
|
|
7 |
Фтор, NiF2/α-Al2O3 , 520 К, 20 сек |
|
|
8 |
470 К, 30 сек |
|
|
9 |
α-AI2O3 (фторированный), 470 К, 30 сек |
|
|
10 |
KJ, 370 К, 1 час |
|
Характерно, что реакционная способность ПФЕН-1, как следует из приведенных данных, существенно ниже, чем у ПФЕН-2, что обусловлено, по-видимому, стерическими особенностями конфигурации их молекул. Различия реакционной способности изомеров перфторнонена наглядно иллюстрируют результаты, полученные при газофазном фторировании их смеси на катализаторе NiF2/α-Al2O3 в недостатке фтора (табл. 6).
Таблица 6. Фторирование смеси ПФЕН-1 и ПФЕН-21 на катализаторе NiF2/α-Al2O3 (температура – 403 К, объемная скорость подачи 140 час-1, мольное отношение азот:ПФЕН = 2,6)
|
№ оп. |
Мольное отношение фтор:ПФЕН |
Степень конверсии, % |
Выход
|
||
|
|
|
F2 |
|||
|
1 |
0,25 |
55,7 |
6,2 |
>99,0 |
98,8 |
|
2 |
0,50 |
80,6 |
28,4 |
>99,0 |
98,4 |
|
3 |
0,80 |
92,8 |
70,1 |
>99,0 |
98,0 |
|
4 |
1,20 |
>99,9 |
>99,9 |
88,4 |
97,4 |
1 Состав исходной смеси, масс.% ![]() -33,2;
-33,2; ![]() -66,2.
-66,2.
Фторирование ПФЕН в слое NiF2/α-Al2O3 при повышенных температурах (420 К и выше) приводит к увеличению образования ПФАО (табл. 4, оп. 13), по-видимому, за счет элиминирования трифторметильной грпуппы у промежуточно образующегося ПФИЛ и последующего селективного фторирования промежуточного ПФЕО с получением ПФАО:
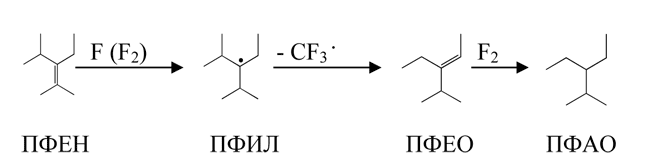
Напомним, что фторирование ПФЕН на известных катализаторах – металлических меди и никеле в этих условиях (температура выше 440 К) протекает неселективно с образованием в основном тетрафторметана и сажи.
Полученные результаты полностью согласуются с основными положениями предлагаемой модели селективного каталитического фторирования. Действительно, локализация фторирования на поверхности катализатора NiF2/α-Al2O3 привела к резкому уменьшению деструктивного фторирования и повышению селективности процесса; отсутствие глубокой деструкции при фторировании в жестких условиях (440 К и выше), столь характерной для фторирования в слое металлических насадок и в полом реакторе, обусловлено, по-видимому, своевременным отводом избыточной энергии промежуточно образующихся возбужденных аддуктов по связям с кристаллической решеткой гетерогенного контакта.
Наличие атомарного фтора на относительно развитой поверхности NiF2/α-Al2O3 (удельная поверхность отработанного катализатора NiF2/α-Al2O3 составляет 2,5-3,0 м2/мл и примерно на три порядка выше, чем у металлических катализаторов) объясняет как резкое увеличение скорости процесса по сравнению с известными катализаторами (это и приводит к локализации фторирования на поверхности), так и особенности селективности фторирования: взаимодействие ПФЕН с молекулярным фтором (в слое инертной насадки и в полном реакторе) стерически затруднено, протекает медленно и останавливается на стадии образования ПФИЛ. Дальнейшее фторирование ПФИЛ возможно только при участии атомарного фтора (благодаря его меньшим геометрическим размерам), что и подтверждается особенностями взаимодействия фтора как с ПФИЛ, так и с ПФЕН в слое NiF2/α-Al2O3.
Природа каталитического действия NiF2/α-Al2O3 при прямом газофазном фторирования ПФЕН обусловлена, таким образом, активацией адсорбированного фтора и, как следствие, локализацией актов фторирования на поверхности; это принципиально отличается от функции металлических насадок, сводящейся к отводу (рассеиванию) тепловой энергии, выделяющейся при фторировании.
С практической точки зрения высокая скорость и селективность прямого газофазного фторирования ПФЕН в слое NiF2/α-Al2O3 позволяет рассматривать этот процесс как основу для эффективной технологии получения ПФАН.
2.3.2 Фторирование перфторгексенов
Одним из возможных путей синтеза 2-перфторметилперфторпентана – эффективной диэлектрической жидкости, является селективное фторирование относительно доступных перфторгексенов – цис- и транс-изомеров 2-перфторметилперфторпентена-2, получаемых анионной димеризацией гексафторпропилена. Данные по прямому газофазному фторированию этих полифторированных олефинов на катализаторе NiF2/α-Al2O3 представлены в табл. 7; здесь же, для сравнения, приведены результаты, полученные с использованием известного металлического никелевого катализатора.
Таблица 7. Прямое фторирование перфторгексенов (температура – 373 К, объемная скорость подачи – 220 час-1, мольное отношение субстрат:фтор:азот = 1:1,2:2,5).
|
# оп. |
Катализатор |
Состав продуктов реакции, объемн.% |
||||||
|
CF4 |
C2F6 |
C3F8 |
C4F10 |
C5F12 |
C6F14 |
C6F12 |
||
|
1 |
Ni, металл |
15,4 |
6,1 |
6,4 |
2,1 |
3,4 |
36,8 |
29,8 |
|
2 |
NiF2/α-Al2O3 |
6,3 |
3,2 |
4,1 |
1,9 |
1,6 |
82,9 |
<0,1 |
Из приведенных данных видно, что прямое газофазное фторирование перфторгексенов на катализаторе NiF2/α-Al2O3 протекает весьма селективно; выход перфторгексана на металлическом никелевом катализаторе существенно ниже.
В случае фторирования перфторгексенов отсутствуют стерические затруднения, поэтому данные для фторирования на испытанных гетерогенных контактах с принципиально различной природой действия, отличаются лишь количественно.
Отметим, что приведенные результаты позволяют рассматривать прямое каталитическое фторирование в качестве перспективного метода для получения 2-перфторметилперфторпентана на основе изомеров 2-перфторметилперфторпентена-2.
2.3.3 Фторирование перфторбутенов
Данные по прямому газофазному фторированию перфторбутенов в смеси с октафторциклобутаном и перфторметилперфторциклобутаном в присутствии плавленого фторида кальция, металлической меди, а также фторида никеля, нанесенного на α-Al2O3, приведены в табл. 8; здесь же для сравнения представлены результаты по фторированию в полом реакторе без катализатора.
Основными продуктами реакции без катализатора являются тетрафторметан и сажа; процесс при этом носит взрывной характер даже при 300 К. Селективного присоединения фтора по двойной связи не происходит; наряду с перфторбутенами деструктивному фторированию подвергаются также октафторциклобутан и перфторметилперфторциклобутан. Последнее согласуется с данными /174/, где показано, что при 550 К и выше фтор реагирует в гомогенных условиях с октафторциклобутаном с образованием энергетически возбужденного промежуточного радикала н-С4F9•, основным продуктом дальнейших превращений которого является тетрафторметан.
Использование насадки из плавленого фторида кальция или металлической меди приводит к некоторому увеличению селективности фторирования (оп. 3-6, табл. 8) – выход перфторбутана достигает 50%. В то же время и в этом случае процесс характеризуется глубокой деструкцией сырья с образованием тетрафторметана.
Принципиальное увеличение селективности фторирования наблюдается при осуществлении процесса в слое NiF2/α-AI2O3 – выход перфторбутана возрастает до 96-99% при степени конверсии префторбутенов, превышающей 99%; октафторциклобутан и перфторметилперфторциклобутан при этом практически не реагируют.
2.3.4. Фторирование гексафторпропилена.
Для оценки эффекта активации фтора поверхностью катализатора на селективность его присоединения по двойной связи в качестве модельной реакции было изучено также газофазное фторирование гексафторпропилена:
С3F6 + F2 = С3F8 –635 кДж/моль
В качестве катализатора был испытан фторид никеля, нанесенный на α-AI2O3 – контакт, обеспечивающий активацию фтора на поверхности и, для сравнения, немодифицированный α-AI2O3, плавленый фторид кальция и известные металлические катализаторы – медь и никель. Часть опытов была проведена в полом реакторе. Данные представлены в табл. 9.
Видно, что взаимодействие фтора с гексафторпропиленом в полом реакторе (даже при разбавлении азотом) протекает неселективно – основным продуктом реакции является тетрафторметан. Заполнение реактора насадкой приводит к специфическому (в зависимости от природы насадки) увеличению вклада селективного фторирования. При использовании фторида никеля, нанесенного на α-AI2O3, (оп. 11,12, табл. 9) фторирование гексафторпропилена протекает наиболее селективно; только в этом случае октафторпропан является основным продуктом даже при использовании неразбавленного фтора. Этот результат является, по-видимому, следствием увеличения вклада селективного гетерогенного фторирования при активации фтора на диспергированном дифториде никеля.
Характерной особенностью прямого фторирования гексафторпропилена является ассоциации промежуточных радикалов с образованием значительных количеств линейного и разветвленного перфторгексанов.
Таблица 9. Прямое фторирование гексафторпропилена.
2.3.5. Фторирование тетрафторэтилена и трифторхлоэтилена
В табл. 10 и 11 приведены сравнительные данные по составу продуктов прямого фторирования тетрафторэтилена и трифторхлорэтилена на катализаторе NiF2/α-Al2O3 и в слое плотной насадки из спиралей тонкой медной проволоки1.
Таблица 10. Прямое фторирование тетрафторэтилена.
|
# |
Катализатор |
Состав продуктов фторирования, объем.% |
|||||
|
CF4 |
C2F6 |
C3F8 |
C4F10 |
C5F12 |
Прочие |
||
|
1 |
NiF2/α-Al2O3 |
24,8 |
34,7 |
12,8 |
12,4 |
4,2 |
11,1 |
|
2 |
Cu, металл |
37,7 |
22,4 |
10,7 |
11,8 |
4,0 |
13,4 |
Из приведенных данных видно, что прямое фторирование тетрафторэтилена и трифторхлорэтилена протекает неселективно с образованием значительных количеств продуктов деструкции и димеризации; выход продуктов селективного фторирования – гексафторэтана и пентафторхлорэтана на NiF2/α-Al2O3 , впрочем, существенно выше, чем при использовании металлической медной насадки.
Таблица 11. Прямое фторирование трифторхлорэтилена.
|
# оп. |
Катализатор |
Состав продуктов фторирования, объемн. % |
|||||||
|
CF4 |
C2F6 |
CF3Cl |
C3F8 |
C2F5Cl |
C4F10 |
C4F8Cl2 |
Прочие |
||
|
1 |
NiF2/α-Al2O3 |
10,6 |
4,2 |
9,6 |
5,7 |
33,6 |
2,5 |
17,3 |
16,5 |
|
2 |
Cu; металл |
18,5 |
4,1 |
13,4 |
7,1 |
20,8 |
2,8 |
16,8 |
16,5 |
Данные по прямому газофазному фторированию гексафторпропилена и, особенно, тетрафторэтилена и трифторхлорэтилена хотя и свидетельствуют о повышении вклада селективного фторирования при использовании разработанного катализатора NiF2/α-Al2O3 (по сравнению с известными металлическими контактами), наглядно иллюстрируют ограничения этого метода. Невысокая селективность фторирования «малых» фторлефинов обусловлена, по-видимому, существенным разогревом реакционных газов при фторировании (вследствие их низкой теплоемкости), когда основной вклад в деструкцию вносит термическое разложение, а не распад энерговозбужденных молекул (как в случае перфторноненов и перфторбутенов).
Интересным в этой связи представлялось изучение прямого каталитического фторирования фторолефинов при разбавлении их инертными, насыщенными фторорганическими соединениями; такое осуществление процесса позволило бы уменьшить разогрев реакционных газов в ходе экзотермического фторирования и, тем самым, снизить вклад термодеструкции.
2.3.6 Двухстадийное фторирование олефинов. Очистка фторхлоруглеродов от олефинов.
Разогрев реакционных газов при прямом фторировании можно уменьшить, осуществив предварительное «мягкое» фторирование олефинов, например, трифторидом кобальта. В этой связи нами было изучено двухстадийное фторирование тетрафторэтилена, ацетилена, гексафторпропилена и трифторхлорэтилена /175/. Отметим, что фторирование ацетилена и гексафторпропилена трифторидом кобальта детально исследовано авторами /176-179/, где, в частности, показано, что выход гексафторэтана при фторировании ацетилена невысок из-за образования значительных количеств продуктов неполного фторирования – пентафторэтана и 1,1,2,2-тетрафторэтана – водородсодержащих аддуктов, дальнейшее фторирование которых на трифториде кобальта протекает относительно медленно. В то же время получение гексафторэтана – ценного рабочего тела для плазмохимической обработки элементов электронных схем, из ацетилена позволило бы осуществить синтез с минимальными затратами фтора без использования промежуточных хлорорганических продуктов. Технология получения тетрафторэтилена, из которого также может быть получен гексафторэтан, включает использование хлороформа, производство которого весьма трудоемко и характеризуется существенными затратами хлора.
В настоящей части работы приведены также данные по прямому каталитическому фторированию:
- тетрахлорэтилена и фтортрихлорэтилена в смеси с фторхлоруглеродами этанового ряда (хладоны-111, -112 и -113); такие смеси образуются при фторировании тетрахлорэтилена гексафторидом урана – наличие в них галоидолефинов усложняет последующую утилизацию хладонов;
- непредельных и водородосодержащих примесей в октафторциклобутане с целью очистки этого перспективного растворителя для сополимеризации тетрафторэтилена и перфторпропилперфторвинилового эфира.
Получение гексафторэтана из ацетилена
Данные по фторированию ацетилена трифторидом кобальта приведены в табл. 12.
Таблица 12. Фторирование ацетилена трифторидом кобальта (скорость подачи ацетилена – 3 л/ч, количество трифторида кобальта – 250г с содержанием активного фтора 13,2 масс.%).
|
# оп |
Тем-пера-тура, К |
Время от начала опыта, час |
Состав реакционных газов, объемн.% |
|||||||
|
CF4 |
C2F6 |
C2H2 |
C2F5H |
CH2F-CF3 |
CHF2- CHF2 |
C4F10 |
Прочие |
|||
|
1 |
473 |
1 |
<0,1 |
0,5 |
1,7 |
25,3 |
7,2 |
62,3 |
<0,1 |
3,0 |
|
2 |
473 |
2 |
<0,1 |
0,3 |
4,8 |
21,8 |
8,8 |
60,1 |
<0,1 |
4,2 |
|
3 |
573 |
1 |
<0,1 |
17,2 |
0,3 |
47,0 |
1,8 |
32,3 |
<0,1 |
1,4 |
|
4 |
573 |
2 |
<0,1 |
6,0 |
4,3 |
46,1 |
3,8 |
38,3 |
<0,1 |
1,5 |
|
5 |
623 |
1 |
0,3 |
72,9 |
0,1 |
22,2 |
0,5 |
1,1 |
1,6 |
0,7 |
|
6 |
623 |
2 |
0,3 |
51,5 |
2,2 |
40,9 |
0,9 |
1,8 |
1,2 |
1,5 |
Из приведенных данных видно, что фторирование в интервале температур 470-625 К протекает практически без деструкции сырья – основными продуктами реакции являются 1,1,2,2-тетрафторэтан, пентафторэтан (при температурах 470-570 К) и гексафторэтан (при 620 К и выше). Характерно, что наряду с продуктом селективного присоединения фтора – 1,1,2,2-тетрафторэтаном, образуются заметные количества 1,1,1,2-тетрафторэтана; аналогичное перераспределение водорода в продуктах фторирования этилена трифторидом кобальта отмечалось авторами /180/.
Уменьшение содержания активного фтора во фториде кобальта, наблюдающееся в ходе процесса, приводит к снижению полноты фторирования (табл. 12, оп. 2, 4, 6).
Из приведенных данных видно также, что даже при 623 К содержание гексафторэтана в реакционных газах не превышает 75 объемн. %; дальнейшее увеличение температуры приводит к интенсивному образованию углерода и тетрафторметана. Отметим, что эти результаты хорошо согласуются с данными по фторированию ацетилена трифторидом кобальта, приведенными в /177/.
В табл. 13 приведены данные по прямому фторированию продуктов взаимодействия ацетилена и трифторида кобальта.
Таблица 13. Прямое фторирование на катализаторе NiF2/α-Al2O3 продуктов взаимодействия ацетилена и трифторида кобальта (объем реактора – 50 мл, скорость подачи: органических продуктов в оп. 1-3 – 2 л/ч, в оп. 4 – 1 л/ч, фтора – 1,5 л/ч).
|
# оп |
Тем-пера-тура, К |
Состав реакционных газов, объемн.% |
||||||||||
|
CF4 |
C2F6 |
C2H2 |
C2F5H |
CH2F- CF3 |
CHF2- CHF2 |
C4F10 |
Прочие |
|||||
|
Состав исходных продуктов |
0,3 |
72,9 |
<0,1 |
22,5 |
0,5 |
1,1 |
1,6 |
0,7 |
||||
|
1 |
473 |
0,3 |
97,0 |
<0,1 |
0,4 |
<0,1 |
0,2 |
1,5 |
0,6 |
|||
|
2 |
523 |
0,3 |
97,4 |
<0,1 |
<0,1 |
<0,1 |
<0,1 |
1,6 |
0,7 |
|||
|
3 |
573 |
0,3 |
97,3 |
<0,1 |
<0,1 |
<0,1 |
<0,1 |
1,6 |
0,8 |
|||
|
Состав исходных продуктов |
<0,1 |
6,0 |
4,3 |
46,1 |
3,8 |
38,3 |
<0,1 |
1,5 |
||||
|
4 |
523 |
25,1 |
72,0 |
<0,1 |
1,4 |
<0,1 |
<0,1 |
<0,1 |
1,5 |
|||
Из приведенных данных видно, что прямое фторирование на катализаторе NiF2/α-Al2O3 продуктов взаимодействия ацетилена и трифторида кобальта сопровождается селективным фторированием пентафторхлорэтана и позволяет получать перфторированные продукты, содержащие
97,0-97,4 объемн. % гексафторэтана (оп. 1-3, табл. 13); фторирование смесей, содержащих значительные количества тетрафторэтана и непрореагировавшего ацетилена, сопровождается образованием тетрафторметана (оп. 4, табл. 13).
Отметим, что таким двухстадийным фторированием тетрафторэтилена, трифторхлорэтилена и гексафторпропилена (сначала трифторидом кобальта, а затем фтором на NiF2/α-Al2O3) могут быть, как показано нами в /175/, практически с количественным выходом получены тетрафторэтан, пентафторхлорэтан и гексафторэтан, соответственно; содержание галоидолефинов в целевых продуктах при этом было ниже чувствительности их определения (0,01 объемн. %).
Данные по прямому фторированию тетрахлорэтилена и фтортрихлорэтилена в смеси с фторхлоруглеродами этанового ряда представлены в табл. 14.
Таблица 14. Прямое фторирование тетрахлорэтилена и фтортрихлорэтилена в смеси с хладонами этанового ряда на катализаторе NiF2/α-Al2O3.*
|
# оп |
Темп., К |
Состав органических продуктов, масс. % |
|||||
|
C2F4Cl2 |
C2F3Cl3 |
C2F2 Cl4 |
C2Cl4 |
C2 Cl3F |
Прочие |
||
|
Состав исход- ных продуктов |
0,3 |
91,8 |
0,3 |
4,8 |
2,6 |
0,2 |
|
|
1 |
333 |
0,3 |
94,3 |
4,6 |
0,4 |
0,1 |
0,3 |
|
2 |
393 |
0,4 |
95,2 |
4,2 |
<0,1 |
<0,1 |
0,3 |
|
3 |
573 |
3,3 |
92,8 |
3,5 |
<0,1 |
<0,1 |
0,4 |
|
Состав исход- ных продуктов |
<0,1 |
24,3 |
60,2 |
12,2 |
0,2 |
0,2 |
|
|
4 |
453 |
0,1 |
27,9 |
71,8 |
<0,1 |
<0,1 |
0,2 |
|
5 |
473 |
0,3 |
28,2 |
71,4 |
<0,1 |
<0,1 |
0,4 |
|
6 |
523 |
0,8 |
31,4 |
67,5 |
<0,1 |
<0,1 |
0,3 |
* Опыты 1-3 проводили в реакторе объемом 50 мл, скорость подачи органических продуктов – 100 г/час, фтора – 2 г/час. Опыты 4-6 проводили в опытном реакторе объемом 6,2л, скорость подачи органических продуктов 10 кг/час, фтора – 0,3 кг/час, азота – 4 м3/час.
Из приведенных данных видно, что фторирование тетрахлорэтилена и фтортрихлорэтилена на катализаторе NiF2/α-Al2O3 протекает селективно с образованием 1,2-дифтортетрахлорэтана и 1,1,2-трифтортрихлорэтана, соответственно, и в интервале температур 390-570 К характеризуется полной очисткой хладонов от нежелательных галоидолефинов; фторирования насыщенных фторхлоруглеродов при этом практически не происходит.
Отметим, что очистка хладонов этанового ряда от тетрахлорэтилена и фтортрихлорэтилена методом прямого каталитического фторирования успешно испытана в промышленном масштабе (на реакторе объемом 60л).
Данные по очистке октафторциклобутана от непредельных и водородсодержащих примесей прямым каталитическим фторированием приведены в таблице 15.
Таблица 15. Очистка октафторциклобутана от непредельных и водородсодержащих примесей прямым фторированием на катализаторе (объемная скорость подачи – 60 час -1, мольное отношение октафторциклобутан:фтор = 20).
|
Наименование примесей |
Содержание примесей в октафторциклобутане, объемн. % |
Содержание примесей объемн.% в октафторциклобутане, очищенном при, К |
|
|
423 |
473 |
||
|
гексафторпропилен |
0,24 |
9х10-4 |
<10-4 |
|
перфторбутин-2 |
6х10-4 |
<10-4 |
<10-4 |
|
2,2-дигидроперфторпропан |
2х10-4 |
10-4 |
<10-4 |
|
2-гидроперфторпропилен |
9х10-4 |
4х10-4 |
<10-4 |
|
транс-перфторбутен-2 |
9,7х10-2 |
<5х10-3 |
<5х10-3 |
|
цис-перфторбутен-2 |
2,1х10-2 |
<5х10-3 |
<5х10-3 |
|
перфторциклобутен |
1,2х10-2 |
<10-4 |
<10-4 |
Из приведенных данных видно, что метод прямого каталитического фторирования позволяет осуществлять тонкую очистку октафторциклобутана от непредельных и водородсодержащих примесей. Очищенный октафторциклобутан, как показали испытания /181/, является эффективным растворителем для осуществления сополимеризации перфторпропилперфторвинилового эфира и тетрафторэтилена с получением фторопласта-50.
Приведенные результаты показывают, таким образом, что разработанный катализатор NiF2/α-Al2O3 позволяет осуществлять прямое селективное фторирование «малых» фторолефинов при их разбавлении инертными насыщенными фторорганическими соединениями. Эти результаты открывают новые практические перспективы для получения чистых фтороуглеродов на основе соответствующих олефинов: их синтез может быть осуществлен в две стадии и включает предварительное фторирование на трифториде кобальта и последующее прямое каталитическое фторирование. Отметим, что первая стадия «мягкого» фторирования может быть заменена рециклом фторуглеродов на каталитическое фторирование.
Разработанный метод может быть использован также для тонкой очистки фторуглеродов от недофторированных соединений.
2.3.7. Фторирование перфторпропионилфторида и низших олигомеров гексафторпропиленоксида. Стабилизация перфторполиэфиров на основе гексафторпропилена и кислорода.
Технология инертных перфторполиэфиров – кислородсодержащих фторуглеродных жидкостей на основе гексафторпропилена и кислорода, широко используемых в специальных областях техники, включает фторирование смеси полимолекулярных продуктов низкотемпературного инициированного окисления, содержащих концевую фторангидридную группу; в результате происходит элиминирование карбонилдифторида и стабилизация перфторполиэфиров /182-184/:
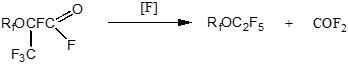
Высокомолекулярные перфторполиэфиры стабилизируют фтором в жидкой фазе, а низкомолекулярные – в газовой фазе трифторидом кобальта; и в том и в другом случае фторирование характеризуется высокой деструкцией ценного фторуглеродного сырья.
В этой связи, а также с целью установления влияния природы гетерогенного контакта на селективность и скорость взаимодействия фторангидридов и фтора, нами были изучены модельные реакции прямого фторирования перфторпропионилфторида и индивидуальных перфторполиэфиров, содержащих концевую фторангидридную группу – низших олигомеров окиси гексафторпропилена: 2-перфторметил-3-оксаперфторгексаноилфторида (димера окиси гексафторпропилена), 2,5-перфтордиметил-3,6-диоксаперфторнонаилфторида (тримера окиси гексафторпропилена) и
2,5,8-перфтортриметил-3,6,9-триоксаперфтордодеканоилфторида (тетрамера окиси гексафторпропилена) – низкомолекулярных аналогов перфторполиэфиров на основе гексафторпропилена и кислорода. Отметим здесь, что прямое газофазное фторирование перфторпропионилфторида и олигомеров окиси гексафторпропилена в полом реакторе, без катализатора, как показали предварительные опыты, носит взрывной неуправляемый характер – основными продуктами этого процесса являются тетрафторметан, карбонилдифторид и сажа. Данные по прямому газофазному фторированию перфторпропионилфторида и низших олигомеров окиси гексафторпропилена в присутствии металлического никеля, α-AI2O3 и катализатора NiF2/α-Al2O3 представлены в табл. 16,17 и на рис. 2.
Таблица 16. Прямое каталитическое фторирование перфторпропинилфторида (объемная скорость подачи 95 час-1, мольное отношение фтор:субстрат = 1,2).
|
# |
Катализатор |
Температура, К |
Степень конвер-сии ПФ1,% |
Состав нейтрализованных реакционных газов2, объмн.% |
||
|
CF4 |
C2F6 |
Прочие |
||||
|
1 |
Ni, металл. |
473 |
14 |
45,6 |
52,3 |
2,1 |
|
2 |
α-AI2O3 |
473 |
22 |
39,4 |
57,1 |
3,6 |
|
3 |
NiF2/α-Al2O3 |
373 |
34 |
35,3 |
60,8 |
3,9 |
|
4 |
То же |
473 |
80 |
20,6 |
79,0 |
0,4 |
1 Степень конверсии перфторпропионилфторида при 373 К на α-Al2O3 и металлическом никеле ниже 1%.
2 Состав нейтрализованных реакционных газов дан без учета диоксида углерода.
Из приведенных в табл. 16 данных видно, что скорость и селективность фторирования перфторпропионилфторида существенным образом зависит от природы применяемого катализатора; наиболее активным является катализатор NiF2/α-Al2O3. Основными продуктами реакции являются гексафторэтан и тетрафторметан; характерно, что при использовании в сопоставимых условиях менее активных катализаторов α-AI2O3 и металичекского никеля относительный выход продукта глубокого деструктивного фторирования – тетрафторметана, существенно выше, чем на NiF2/α-Al2O3.
Из приведенных в табл. 17 и на рис. 2 данных видно, что природа гетерогенного контакта оказывает существенное влияние на скорость фторирования низших олигомеров окиси гексафторпропилена.
Так, степень конверсии тримера окиси гексафторпропилена на α-AI2O3, металлическом никеле и NiF2/α-Al2O3 при 473 К составляет 11, 35 и 90 %, соответственно (оп . 10,13,16, табл. 17). Обращает внимание практически количественный выход 5,8-перфтордиметил-3,6,9-триоксаперфтордодекана при фторировании тетрамера окиси гексафторпропилена на катализаторе NiF2/α-Al2O3 (оп. 17, табл. 17).
Каталитическое газафазное фторирование низших олигомеров окиси гексафторпропилена может включать образование неустойчивых перфторалкилгипофторитов, последующая деструкция которых, в соответствии с /185/, протекает с элиминированием карбонилдифторида и образованием стабильных фторэфиров с концевой перфторэтоксильной группой. Это предположение согласуется с отмеченной ранее повышенной активностью катализатора NiF2/α-Al2O3 при синтезе трифторметилгипофторита из оксида углерода или карбонилдифторида.
Из приведенных в табл. 17 даных также видно, что реакционная способность низших олигомеров окиси гексафторпропилена неодинакова – так, при 473 К степень конверсии димера, тримера и тетрамера на катализаторе NiF2/α-Al2O3 составила 52, 90 и 98 %, соответственно. Увеличение реакционной способности фторангидридов с ростом их молекулярной массы весьма важно с практической точки зрения и свидетельствует о повышенной реакционной способности относительно высокомолекулярных перфторполиэфиров, образующихся при низкотемпературном инициированном окислении гексафторпропилена. Газофазное фторирование высокомолекулярных перфторполиэфиров, впрочем, возможно только при пониженном давлении или разбавлении инертным газом.
Таблица 17. Прямое каталитическое фторирование низших олигомеров гексафторпропиленоксида (объемная скорость подачи – 95 час-1, мольное отношение фтор:субстрат = 1,2).
|
# оп. |
Катализатор |
Температура реакции, К |
Степень конверсии органического субстрата % |
Выход основного продукта,1 % |
|
1. Фторирование димера ОГФП |
||||
|
1 |
Ni, металл. |
423 |
9 |
97 |
|
2 |
То же |
473 |
15 |
91 |
|
3 |
α-AI2O3 |
423 |
3 |
98 |
|
4 |
То же |
473 |
7 |
98 |
|
5 |
NiF2/α-Al2O3 |
393 |
14 |
99 |
|
6 |
То же |
423 |
28 |
99 |
|
7 |
- «- |
473 |
52 |
98 |
|
2. Фторирование тримера ОГФП |
||||
|
8 |
Ni, металл. |
393 |
6 |
98 |
|
9 |
То же |
423 |
16 |
98 |
|
10 |
-«- |
473 |
35 |
92 |
|
11 |
α-AI2O3 |
393 |
2 |
99 |
|
12 |
То же |
423 |
4 |
99 |
|
13 |
-«- |
473 |
11 |
98 |
|
14 |
NiF2/α-Al2O3 |
393 |
22 |
99 |
|
15 |
То же |
423 |
53 |
99 |
|
16 |
-«- |
473 |
90 |
98 |
|
3. Фторирование тетрамера ОГФП |
||||
|
17 |
NiF2/α-Al2O3 |
473 |
98 |
96 |
1 Основными продуктами селективного фторирования димера, тримера и тетрамера ОГФП явяются 3-оксаперфторгексан, 5-перфторметил-3,6-диоксаперфторнонан и 5,8-перфтордиметил-3,6,9-триоксаперфтордодекан, соответственно. Выход основных продуктов определяли в расчете на прореагировавший субстрат.
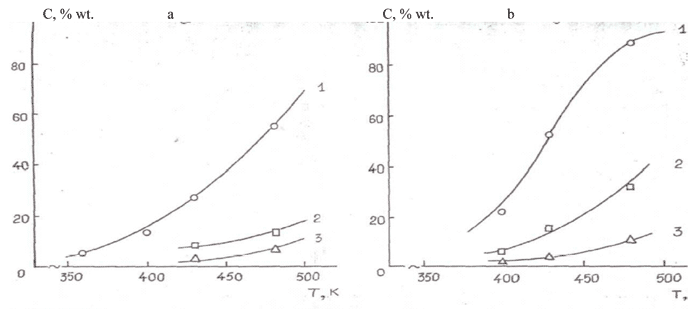
Рисунок 2. Температурные зависимости концентраций 3-оксаперфторгексана (а) и 5-перфторметил-3,6-оксаперфторнонана (b) в жидких продуктах прямого газофазного фторирования димера и тримера гексафторпропиленоксида, соответственно, на катализаторах: 1 – NiF2/α-Al2O3; 2 – металлическом никеле и 3 – α-AI2O3.
Эффективный катализатор фторирования низших олигомеров окиси гексафторпропилена NiF2/α-Al2O3 был испытан нами для газофазной стабилизации легкой фракции перфторполиэфиров на основе гексафторпропилена и кислорода (температура выкипания 313-343 К при 133Па). Технология стабилизации этой фракции в настоящее время основана, как уже отмечалось, на фторировании трифторидом кобальта и характеризуется низкой производительностью, повышенной материало- и энергоемкостью.
Сравнительные данные по фторированию перфторполиэфиров фтором на катализаторе NiF2/α-Al2O3 и трифторидом кобальта приведены в табл. 18.
Таблица 18. Стабилизация легкой фракции перфторполиэфиров на основе гексафторпропилена и кислорода.
|
# оп. |
Температура, К |
Потери сырья при фторировании, масс. % |
Остаточное содержание отщепляемого фториона1, масс. % |
Степень фторирования % |
|
1. Прямое фторирование на катализаторе NiF2/α-Al2O32 |
||||
|
1 |
573 |
7,1 |
1,0 х 10-1 |
91,3 |
|
2 |
623 |
8,5 |
8,2 х 10-3 |
99,3 |
|
3 |
643 |
9,7 |
4,3 10-3 |
99,6 |
|
4 |
673 |
10,6 |
8,0 х 10-4 |
>99,9 |
|
2. Фторирование трифторидом кобальта3 |
||||
|
5 |
573 |
16,3 |
2,5 х 10-1 |
78,3 |
|
6 |
623 |
18,7 |
1,4 х 10-1 |
87,8 |
|
7 |
673 |
20,8 |
5,5 х 10-2 |
95,2 |
1 Содержание фторангидридных групп в перфторполиэфирах оценивали по концентрации фтор-иона, отщепляемого при длительном термическом водно-щелочном гидролизе; его содержание в исходных эфирах составило 1,15 масс. %.
2 Количество катализатора – 50мл, скорость подачи эфиров – 50г/час, фтора – 4 г/час, степень конверсии фтора при 573, 623, 643 и 673 К составило 44, 51, 58 и 65 %, соответственно.
3 Количество трифторида кобальта – 250г, содержание активного фтора – 13,4 масс.%, скорость подачи эфиров – 50 г/час.
Приведенные данные наглядно иллюстрируют преимущества каталитического метода, которые заключаются в высокой скорости и селективности фторирования. Потери сырья при каталитическом фторировании в весьма жестких условиях (673 К) составили всего
10,6 масс. % (оп. 4, табл. 18), что существенно ниже, чем при использовании в аналогичных условиях трифторида кобальта (20,8 масс. % - оп. 7, табл. 18). Различие в скоростях фторирования можно оценить по остаточному содержанию непрореагировавших фторангидридов (определяли по
концентрации отщепляемого фтор-иона во фторированных эфирах) – их количество в продуктах прямого каталитического фторирования более чем в 50 раз ниже, чем при использовании трифторида кобальта (оп. 4,7 табл. 18).
продолжение в следующем выпуске
Список литературы
2. Теддер Д.М. Фторирование органических соединений элементарным фтором. В кн."Успехи химии фтора", т. I-II -М.-Л. : Химия, 1964, с. 380-423.
3. Оркин В.Л., Чайкин A.M. Определение констант скорости образования атомов в реакциях молекулярного фтора с окисью азота, этиленом и тетрафторэтиленом. Кинетикаикатализ, 1982,т.23,в.З,с.529-533.
4. Anson P.O., Fredricks P.S., Tedder J.M. Free-radical substitutionin aliphatic compounds. Part 1. Halogenation of n-butahe and isobutane in the gas phase. J.Chem.Soc, 1959, March, p.918-922.
5. Fredricks P.S., Tedder. J.M. Free-radical substitution in aliphatic compounds. Part 11. Halogenation of the n-butylhalides. -J.Chem.Soc, 1960, Jan., p. 144-150.
6. Purington S.T., Kagen B.S., Patric T.B. The application of elemental fluorine in organic synthesis. Chem.Rev., 1986, № 86, p.997-1018.
7. Aikman R.E., Lagow E.J. Syhthesis of tetra-cis-(perfluorocyclohexyl)-methane and bis-(perfluorocyclohexyl)-methane by direct fluorination. -J.Org.Chem., 1982, v.47, № 14, p.2789-2790.
8. Еременко Л.Т., Орешко Г.В. Фторирование элементарным фтором лабильных полинитросоединений – аддуктов реакции Михаэля. -Изв. АН СССР, сер. Химия, 1969, № 2, с.479.
9. Еременко Л.Т., Нацибуллин Ф.Я., Боровинская Й.П., Карпова Н.Д. Синтез перфторнитроэтанов. -Изв. АН СССР. Сер.хим., 1968, № 2, с.429-430.
10. Патент США 4523039. Способ получения перфорированных простых эфиров. /Лагов Р.Дж., Герхарт Дж.Е. -Опубл. 11.06.85, РЖ Химия, 1986, 7Н36.
11. Патент США 3242218. Способ получения фторуглеродных полиэфиров. /Миллер В.Т. -Опубл.22.03.66,РЖХимия, 1967, 12C232.
12. Des Martean D.D, Fluoroperoxytrifluoromethane CF300F. Preparation from trifluoromethyl hydroperoxide and fluorine in the presence of cesium fluoride. -Inorg Chem., 1972, v.11, № 1, p.193-195.
13. Merritt R.F., Johnson F.A. Direct fluorination. Addition of fluorine to indenes and.acenaphthylenes, -J.Org.Chem., 1966, v.31. № 6, p.1859-1863.
14. Merritt R.F, Johnson F.A. Direct fluorination of steroidal olefines to cis-vicinal difluorides. –J.Am.Chem.Soc., 1966, v.88, № 8, p.1822-1823.
15. Патент США.3487093. Фторированные олефины. Меррит Р.Ф. –Опубл.30.12.69,РЖХимия, 1970, 23Н29.
16. Grakauskas V. Direct liquid phase fluorination of halogenated aromatic compounds. -J.Org.Chem., 1969, v.34, № 10, p.2835-39.
17. Brooke G.M., Chambers B.D., Heyes J., Musgrave W.K.R. Direct preparation and some reactions ofсhlorofluorobenzenes. -J.Chem.Soc., 1964, Febr., p.729-735.
18. Merritt R.F, The polar addition of molecular fluorine to acetylenes. –J.Org.Chem., 1967, v.32, № 12, p.4124-4126.
19. Шеппард У., Шартс К. Органическая химия фтора. -
М.: Мир, 1972, с.52, 89, 112.
20. Миллер В., Эренфельд Дж, Фелан Дж., Пробер М., Рид Ш, Фторирование полностью галоидированных олефинов.«Химияфтора», № 2, -М.:ИЛ, 1950,с.228-240.
21. Miller W.T. My early days in fluorine chemistry. –J.Fluor.Chem., 1981, v.18, p.305-321.
22. Miller W.T., Stoffer J.O., Fuller G., Currie A.C. The mechanism of fluorination. IV. The effect of temperature and of fluorine concentration on reaction. A new fluorination apparatus. –
J.Am.Chem.Soc., 1964, v.86, № 1, p.51–56.
23. Исикава Нобуо, Китацумэ Томоя. Новые методы фторирования ароматических соединений. –Юкки госэй качаку кёкайси., 1976, т.34, № 3, с.173-178, РЖ Химия, 1976, 23Ж360.
24. Патент Японии 55-18695. Способ фторирования./ Маруо Кэйити, Мисаки Сусуму. –Опубл. 21.05.80, РЖ Химия, 1981, 5
Н122.
25. Sirip L.A., Lagow R.J. Direct fluorination of 2,2,4,4-tetramethyl pentane. Sterically protected residual protons. –J.Org.Chem., 1977, v.42, № 21, p.3437-3438.
26. Kowanko N., Branthaver J.F., Sugihara J.M. Direct liquid-phase fluorination of petroleus. –Fuel, 1978, v.57, № 12, p.769-775.
27. Патент США 40004996. Фторирование органических соединений./ Коллониц Дж. –Опубл. 25.01.77, РЖ Химия, 1977, 23Н57.
28. Патент Великобритании 1077065. Фторирование нитросоединений./ Гракаускас В., Хэмел Э.Э. –Опубл. 26.07.67, РЖ Химия, 1975, 16Н84.
29. Merritt R.F. Direct fluorination of 1,1-diphenylethylene. –J.Org.Chem., 1966, v.31, № 11, p.3871-3873.
30. Merritt R.F. The polar fluorination of propenylbenzene. –J.Am.Chem.Soc., 1967, v.89, № 3, p.609-612.
31. Merritt R.F., Johnson F.A. Low-temperature fluorination of Schiff bases. -J.Org.Chem., 1967, v.32, № 2, p.416-419.
32. Misaki S. Direct fluorination of phenol and cresols. –
J.Fluor.Chem., 1981, v.17, № 2, p.159-171.
33. Maxwell A.F., Detoro F.E., Bigelow L.A. The action of elementary fluorine upon organic compounds. XXIII. The jet fluorination of certain aliphatic hydrocarbons as oriented and controlled by operation conditions. -J.Am.Chem.Soc., 1960, v.82, № 22, p.5827-5830.
34. Attaway J.A., Groth R.H., Bigelow L.A. The action of elementary fluorine upon organic compounds. XXIII. The fluorination of some amides, nitriles and of methylthiocyanate. -J.Am.Chem.Soc., 1959, v.81, № 14, p.3599-3603.
35. Robson P., Mc Longhlin V.C.R., Hynes J.B., Bigelow L.A. The action of elementary fluorine upon organic compounds. XXIV. The jet fluorination of hydrogen cyanide, cyanogen, methylamine and ethylenediamine. Pyrolysis and fluorinolysis of selected products. -J.Am.Chem.Soc., 1961, v.83, № 24, p.5010-5015.
36. Boffenberg K. Substitution von benzol durch elementares Fluor in der Gasphase. –Chem.Ztg.,1972, v.96, № 2, p.84-92.
37. Патент Японии 58-41829. Получение октафторпропана./ Фукуи Сиро, Йонэда Хадзимэ. –Опубл.11.03.83,РЖХимия, 1984, 5Н11.
38. Hayes L.J., Dixon D.D. Direct fluorination of polyester and related compounds. –J.Fluor.Chem., 1977, v.10, № 1, p.1-16.
39. Gerhardt G.E., Lagow R.J. Synthesis of perfluoropolyethers by direct fluorination: a novel preparation for perfluoro (polypropylene oxide) ethers and perfluoro (polymethylene oxide) ethers. –J.Chem.Soc., 1981, part 1, № 5, p.1321-1328.
40. Патент США 4113772. Метод получения олигомеров перфторэфиров с концевыми карбоксильными группами. / Лагов Р.Дж. –Опубл.12.09.78,РЖХимия, 1979, 15Н14.
41. Gerhardt G.E., Lagow R.J. Synthesis of perfluoropoly (ethylene glycol) ethers by direct fluorination. –J.Org.Chem., 1978, v.43, № 23, p.4505-4509.
42. Adcock J.L., Znoue Shoji, Lagow R.J. Simultaneous fluorination and functionalization of hydrocarbon polymers. –J.Amer.Chem., 1978, v.100, № 6, p.1948-1950.
43. Lagow R.J., Margrave J.L. The controlled reaction of hydrocarbopolymers with elemental fluorine. –J.Polym.Sci.: Polym.Lett.Ed., 1974, v.12, № 4, p.177-184.
44. Pat. 3775489 (USA). Process for fluorination of aromatic and polynuclear hydrocarbon compounds and fluorocarbons produced thereby. Margrave J.L., Lagow R.J. –Publ. 27.11.73.
45. Lagow R.J., Maraschin N.J. Direct fluorination of cyclic and bicyclic hydrocarbons. –7th Int.Symp.Fluorine Chem., Santa Cruz, Calif., 1973, s.l., s.a., p.1-25.
46. Gerhardt G.E., Dumitru E.T., Lagow R.J. Synthesis of hightbranched perfluoroethers by direct fluorination, promising new materials based on the hexafluoroacetone –ethylene copolymer. –J.Polym.Sci., 1980, v.18, № 1, p.157-169.
47. Robertson G., Liu E.K.S., Lagow R.J. Synthesis of perfluoroadamantane compounds by direct fluorination. –J.Org.Chem., 1978, v.43, № 26, p.4981-4983.
48. Lagow R.J. Large scale synthesis of organofluorine compounds using elemental fluorine; a third Simons cell. –Int.Symp. “Centenary of the discovery of fluorine”, Abstr., Paris, 1986, p.2.
49. Adcock J.L., Lagow R.J. The synthesis of the fluorinated ethers “perfluoroglyme” and “perfluorodiglyme” by direct fluorination. –J.Org.Chem., 1973, v.38, № 20, p.3617-3618.
50. Koshar R.J., Hausted D.R., Meiklejohn R.A. Organic fluoronitrogens. V. Bis(difluoroamino)difluoromethane. –
J.Org.Chem., 1966, v.31, № 12, p.4232-4234.
51. Koshar R.J., Hausted D.R., Wright C.D. Organic fluoronitrogens. VII. Tris(difluoroamino)fluoromethane and related compounds. –J.Org.Chem., 1967, v.32, № 12, p.3859-3864.
52. Патент США 3981783. Процесс электрохимического фторирования с дополнительной подачей водорода и увеличением выхода по току. / Чайлдс В.В. –Опубл.21.09.76, РЖ Химия, 1977, 14Л237.
53. Патент Германии 2106870. Способ электрохимического фторирования органических соединений./Восс П., Нидерпрум Х., Кауль Г., Трепп Р., -Опубл. 24.02.77, РЖ Химия, 1977, 24Н16.
54. Патент Германии 2302132, 1976. Способ получения разветвлённых перфторалканов. /Беннингер С. -Опубл. 23.12.76, РЖ Химия, 1977, 24Л202.
55. Патент США 4035250. Способ получения перфторгептана. /Вальтерс Х.С., Чайдс В.В. -Опубл.12.07.77, РЖ Химия, 1978, 6Н22.
56. Патент Японии 53-18488. Способ получения перфторциклоалканов. /Сато Дайсукэ, Ямамути Коити, Мурасима Рёширо. -Опубл. 15.06.78, РЖ Химия, 1979, 9Н121.
57. Патент США 3662009. Получение ненасыщенных фторсодержащих соединений. /Хадчинсон В.М. -Опубл. 09.05.72, РЖ Химия, 1973, 9Н12.
58. Гольдштейн Б.В., Серушкин И.Л., Никонорова Н.И. О роли фторида никеля в реакциях электрохимических соединений. –Тр. ГИПХ, № 39, часть 1, инв. № Т-1863., Л., 1975, с.43-47.
59. Фаулер Р.Д., Берфорд Н.Б., Гамильтон Дж.М., Свит Р.Дж., Уэбер К.Е., Каспер Дж.С. Лайтайн Н. Синтез фторуглеродов. В кн. «Химия фтора», сб. № 1 –М.: ИЛ, 1950, с.91-113.
60. Беннер Р.Дж., Беннинг А.Ф., Доунинг Ф.Б., Ирвин С.Ф., Джонсон К.С., Линч А.Л., Пармели Х.М., Уирт У.У. Фторуглероды, полученные фторированием углеводородов трехфтористым кобальтом. В кн. «Химия фтора», сб. № 1 –М.: ИЛ, 1950, с.114-128.
61. Фаулер Р.Д., Андерсон Х.С., Гамильтон Дж.Н., Берфорд Н.Б., Спагетти А., Битердих С.Б., Лайтайн Н. Фториды металлов, применяемые в синтезе фторуглеродов. В кн. «Химия фтора», сб. № 1 –М.:ИЛ, 1950, с.143-153.
62. Патент Японии 60-81134. Получение октафторпропана. /Катамура Коити, Кагэяма Ютака, Накаяма Хидэтоси. –Опубл. 09.05.85, РЖ Химия, 1986, 12Н25.
63. Патент Японии 60-10933. Получение гексафторэтана./ Катамура Коити, Кагэяма Ютака, Накаяма Хидэтоси. –Опубл. 15.06.85, РЖ Химия, 1986, 11Н21.
64.PakV.,PekaJ.,CermakV.,SykoraF.,PetrzilaV.Poloprovoznizarizeniprovyrobuperfluororganickychlatek.
–Chem.Prum., 1978,v.28, № 9,p.467-470.
65. Патент Франции 2381732. Усовершенствованный способ перфторирования циклических углеводородов. / Мур Р.Е. –
Опубл. 27.10.78, Изобретения в СССР и за рубежом, 1979, в.55, № 5, с.67.
66. Патент США 4143079. Способ получения перфтор-1-метил-4-изопропилциклогексана из пинена. / Мур Р.Е. –Опубл. 06.03.79, Изобретения в СССР и за рубежом, 1979, в.55, No 21, с.20.
67. Патент Великобритании 1597914. Способ перфторирования циклических углеводородов. / Сантэч инкорп. –Опубл. 16.09.81, Изобретения в СССР и за рубежом, 1982, в.57, № 12, с.64.
68. Burdon J. The exhaustive fluorination of aliphatic compounds.
–Int. Symp. “Cent. ...”, Paris, 1936, p.9.
69. Moore R. E., Driscoll G. Perfluorination of bicyclic and tricyclic hydrocarbons. –IV-th Winter Fluorine Conf., Abstr, Daytona Bearen, 1979, p.8.
70. Асович В.С., Прокудин И.П. Особенности фторирования органических веществ четырехфтористым церием. –Тр. ГИПХ, № 40, инв. № Т-2117, Л., 1976, с.25-31.
71. Исикава Н., Кобаяси Ё. Фтор. Химия и применение. –
М.: Мир, 1982, с.91.
72. Кэйджи Дж..Х., Гроссе А.В., Барбер Е.Дж., Бэрджэр Л.Л., Шелдон З.Д. Получение фторуглеродов каталитическим фторированием углеводородов. В кн. «Химия фтора», сб. № 1 –М.: ИЛ., 1950, с.129-135.
73. Бигелоу Л.А. Действие элементарного фтора на органические соединения. В кн. «Фтор и его соединения (под ред. Дж. Саймсона), т.1 –М.: ИЛ., 1953, с.314-335.
74. Musgrave W. K. R., Smith F. Organic Fluorides. PartІІ. The effect of metals on the fluorunationof hydrocarbons. –J. Chem. Soc., 1949, p.3026-3028.
75. Брик Т. Дж. Фторуглероды; их свойства и производство во время войны. В кн. «Фтор и его соединения» (под ред. СаймонсаДж.),т.І –М.:ИЛ, 1953,с.355-388.
76. Maraschin N. J., Catsikis B. D., Davis L. H., Larvinen J., Lagov R. J. Synthesis of structurally unusual fluorocarbons by direct fluorocarbon. –J.Am. Chem. Soc., 1975, v. 97, № 3, p.513-517.
77. Патент США 4113453. Аппарат для низкотемпературного прямого фторирования. / Лагов Р.Дж, Адкок И.Л., Марашин Н.И. –Опубл. 12.09.78, РЖ Химия, 1979, 19И130.
78. Патент США 4281119. Аппарат для прямого фторирования с контролируемым охлаждением по зонам. / Лагов Р.Дж., Адкок И.Л., Марашин Н.И. –Опубл. 28.07.81, РЖ Химия, 1982, 9Н225.
79. Рахимов А.И., Химия и технология фторорганических соединений. –М.: Химия, 1986, с.9-10.
80. Schmeisser M., Ehlers K. P., Sartori P. Dichlorhexafluorpropan durch fluorierung von 1,2-dichlorpropan. –Angew. Chem., 1967, v.79,№ 13, p.622.
81. Margrave W. K. R., Smith F. Organic Fluorides. PartІ. Fluorination of hydrocarbons. –J. Chem. Sоc., November, 1949, p.3021-3026.
82. Гроссе А.В., Кэйди Дж. Х. Свойства фторуглеродов. В кн. «Химия фтора: сб. № 1 –М.: ИЛ, 1950, с.35-64.
83. Patent GB 1281822. Improved Fluorination Process. / Kingdom R. J., Bond G. D. –Publ. 19.07.72.
84. Hill M. Process and market development of fluorocarbon fluids. –Chem.аnd Ind., 1975, № 3, p.118-121.
85. Патент Франции 2028457 (В). Способ фторирования. / Империал Смелтинг Корп. –Опубл. 15.01.70, Офф. Бюлл. Франции, Химия и металлургия, 1970, № 45-48, ч. І, с.44.
86. Патент США 4220606. Способ получения перфторпроизводных из цикличеких углеводородов. / Мурр Р.Е. –Опубл.02.09.80,РЖХимия, 1981, 9Н105.
87. Patent US 3480667. Method of producing fluorinated compounds. / Siegart W. R., Blackley W. D. –Publ. 25.11.69.
88. Патент США 4330475. Аэрозольный способ прямого фторирования и устройство для этой цели. / Адкок Д.Л., Рэнк Е.Б. –Опубл.18.05.82,РЖХимия, 1983,ІІН76.
89. Ruff J. K. The catalytic fluorination of perfluorocarbon nitriles and imines. –J. Org. Chem., 1967, v.32,№ 5, p.1675-1677.
90. Lusting M., Ruff J. K. Fluorination of some perfluoro alkyliminosulfurdifluorides. –Inorg. Chem., 1965,v.4, № 10,p.1444-1446.
91. Фокин А.В., Столяров В.П., Радченко В.П. Синтезполифторамино-и α-фторнитросоединений на основе реакций газообразного фтора. –Изв. АН СССР, сер. хим., 1982, № 8, с.1853-1861.
92. Фокин А.В., Галахов В.С., Узун А.Т. и др. Реакция фтора с солями щелочных металлов динитроацетонитрила в присутствии фторидов калия или кальция. –Изв. АН СССР, сер. хим., 1974, № 2, с.456-458.
93. Фокин А.В., Узун А.Т., Столяров В.П. Бис (дифторамино) фторацетальдегид –новый представитель α, α-бис (дифторамино) альдегидов. –Изв. АН СССР, Сер. хим., 1982, № 6, с.1438.
94. Еременко Л.Т., Нацибуллин Ф.Я., Боровинская И.П., Карпова Н.Д. Синтез перфторнитроэтанов. –Изв. АНСССР,Сер.хим., 1968, № 3,с.431-432.
95. Sekiya A., Des Martean D. D. Synthesis of 1,1-bis (fluoroxy) –perhaloalkanes by reaction of fluorinated carboxylic acids with fluorine in the presence of cesium fluoride. –Inorg. Chem., 1980, v. 19, № 5, p.1328-1330.
96. Lu S., Des Martean D. D. Direct synthesis of fluorinated peroxides. 7. Perfluoro-tret-butyl-fluoroformyl peroxide. –Inorg. Chem., 1978, v. 17, № 2, p.304-306.
97. Schack C. J. A new synthesis of difluoraminotrifluoromethane. –J. Fluor. Chem., 1981,v. 18,no 4,p.583-586.
98. Патент Германии 2712732. Способ получения октафторпропана. /Халац С.П. –Опубл. 28.09.75, РЖ Химия, 1979, 14Н14.
99. Патент США 4158023. Способ получения октафторпропана. / Халац С.П. –Опубл. 12.06.79. Изобретения в СССР и за рубежом, 1979, в. 55, № 24, с.121.
100. Патент Великобритании 1568020.Способ получения октафторпропана. / Халац С.П. –Опубл. 21.05.80. Изобретения в СССР и за рубежом, 1981, в. 55, № 3, с.54.
101. Патент США 4377715. Получение перфторпропана. /Нучка Х.Р., Хино И.Б., Эйбэк Р.Е., Робинсон Н.А. –Опубл. 22.03.83, Изобретения в СССР и за рубежом, 1983, в.57, № 23, с.88.
102. Европейский патент 0031519. Способ фторирования органических соединений элементарным фтором. / Эллайд Кэмикэл Ко. –Опубл. 08.07.81, Изобретения в СССР и за рубежом, 1983, в.57, No 1, с.20.
103. Европейский патент 0032210. Способ фторирования органических соединений фтором в трубчатом реакторе из пористого металла в присутствии перфторированного разбавителя. / Эллайд Кэмикл Ко. –Опубл. 22.07.81, Изобретения в СССР и за рубежом, 1983, в.57, № 2, с.27.
104. Патент США 4513154. Способ проведения последовательно-конкурирующих газофазных реакций. / Курц Б.Е. –Опубл. 23.04.85, РЖ Химия, 1986, 4Н25.
105. Патент США 2831035. Производство фторированных углеродов. / Тучковский Э.А., Вульф К. –Опубл. 15.04.58, РЖ Химия, 1960, 85740.
106. Патент США 3709800. Получение перфторированных углеводородов./ Фокс Х.М. –Опубл. 09.01.73, РЖ Химия, 1973, 22Н23.
107. Патент Японии 5608. Фторсодержащие галоидуглеводороды. / Осиба Такаси. –Опубл. 22.03.65, РЖ Химия, 1968, 5Н35.
108. Патент США 2681267. Процесс улучшения каталитических свойств фтористого алюминия и продуктов из него. / Калфи Дж. Д., Миллер Ч.Б. –Опубл.15.06.54,РЖХимия, 1956, № 14, 45553.
109. Hohorst F. A., Shreeve J. M. Bis-(fluoroxy)-difluoromethane,CF2(OF)2. –J. Am. Chem. Soc., 1967, v.89, № 8, p.1809-1819.
110. Walker N., Des Martean D. D. Direct Synthesis of fluorocarbon peroxides.ІІІ. The addition of chloroperoxytrifluoromethane to olefins. –J. Am. Chem.Soc., 1975, v.97,№ 1, p.13-17.
111. Патент США 4499024. Непрерывный способ получения бис (фторокси) дифторметана. / Филолт М.Ж. –Опубл.12.02.85,РЖХимия, 1985, 20Н14.
112. Lusting M. A., Pitochelli A. R., Ruff J. K. The catalytic addition of fluorine to a carbonyl group. Preparation of fluoroxy compounds. –J. Am. Chem. Soc., 1967, v. 89, № 12, p.2841-2843.
113. Ruff J. K., Pitochelli A. R., Lusting M. A. A simple synthesis of fluoroxyperfluoroalkyl compounds. -J. Am. Chem. Soc., 1966, v.88,№ 19, p.4531-4532.
114. Kennedy R. C., Cady G. H. Reaction of carbonyl fluoride with fluorine in the presence of various fluorides as catalysts. –J. Fluor. Chem., 1973,v.3, № 1, p.141-154.
115. Мухаметшин Ф. М. Успехи химии фторорганических гипогалогенитов и родственных соединений. –Успехи химии, 1980, т. 49, № 7, с.1260-1288.
116. Мухаметшин Ф.М. Гипофториты и их применение в органическом синтезе. –В кн. «Новые фторирующие реагенты в органическом синтезе». –Новосибирск: Наука, 1987, с.140-196.
117. Cady G. H. Proposed mechanisms of catalysis of fluorination of CF2O. –Anales. Asoc. Quim. Argentina, 1971, v.59, p.3-4, p.125-131.
118. Patent US 3230264. Reaction of carbonyl fluoride with fluorine. –Roger S. P., Cady G.H. –Publ. 18.01.66.
119. Wechsberg M., Cady G.H. Comparative studies of catalytic fluorination of carbon monoxide with elementary fluorine. –J. Am. Chem. Soc., 1969, v.91, p.4432-4435.
120. Kellog K. B., Cady G.H. Trifluoromethyl hypofluorite. –J. Am. Chem. Soc., 1948, v.70,№ 12, p.3986-3988.
121. Gervasi J.A., Brown M., Bigelow L. A. The action of elemental fluorine upon organic compounds. XX. The fluorination of mono-, di-and trimethylamine, ethylenediamine and ethyleneimine. –J. Am. Chem. Soc., 1956, v.78, № 8, p.1679-1682.
122. Avonda F. P., Gervasi J. A., Bigelow L.A. The action of elemental fluorine upon organic compounds. XXІ. The fluorination of malononitrile and dimethylformamide. –J. Am. Chem. Soc., 1956, v.78,№ 12, p.2798-2800.
123. Тойтельбойм М.А., Шойхет А.А., Каплунов М.Г., Веденеев В.И. Энергетические разветвления цепей в реакциях трифторметилгипофторита с галоидметанами. –Кинетика и катализ, 1981, т. 22, в.2, с.298.
124. Тойтельбойм М.А., Шойхет А.А., Веденеев В.И. Энергетические разветвления цепей в реакциях трифторметилгипофторита с галоидметанами. 2. Механизм отрицательного взаимодействия цепей. –Кинетика и катализ,
1981, т.22, в.3, с.564-568.
125. Веденеев В.И., Парийская А.В. Механизм фторирования метана и его фторпроизводных. І. Сравнение скоростей фторирования метана, фторметана, дифторметана и трифторметана. –Кинетика и катализ, 1971, т.12, в.1, с.21-26.
126. Парийская А.В., Веденеев В.И. Механизм фторирования метана и его производных. 2. Дифторметан –Кинетика и катализ, 1971, т.12, в.2, с.293-298.
127. Парийская А.В., Веденеев В.И. Механизм фторирования метана и его фторпроизводных. 3. Фтористый метил. –
Кинетика и катализ, 1971, т.12, в.3, с.543-548.
128. Парийская А.В., Веденеев В.И. Механизм фторирования метана и его фторпроизводных. 4. Метан. –Кинетика и атализ, 1971, т. 12, в.4, с. 839-842.
129. Надточенко В.А., Федотов Н.Б., Веденеев В.И., Саркисов О.М. О реакции колебательно возбужденной молекулы СН3F с фтором. –Докл. АН СССР, 1978, т.238, № 6, с.1391-1394.
130. Парийская А.В., Веденеев В.И. О природе задержек самовоспламенения в системе СН3F + F2 + O2 + Не. –Кинетика и катализ, т.14, в.6, с.1365-1369.
131. Веденеев В.И., Тейтельбойм М.А., Шойхет А.А. Фотохимическое фторирование фтороформа в присутствии кислорода. –Изв. АН СССР, 1976, № 9, с.1968-1970.
132. Медведев Б.А., Тейтельбойм М.А., Шилов А.Е. Химическая активация солекулы СHFCl2 в реакции СНCl2+ F2→ СНСl2F + F . –Кинетика и катализ, т.12, в.2, с.269-275.
133. Медведев Б.А., Тейтельбойм М.А., Шилов А.Е. Механизм газофазной реакции хлорфторметана с молекулярным фтором. –Кинетика и катализ, 1971, т.12, в.3, с.49-751.
134. Веденеев В.И., Медведев Б.А., Тейтельбойм М.А. Газофазное фторирование дифторметана при повышенных давлениях инертного газа. –Кинетика и катализ, т.13, в.1, с.50-53.
135. Обвивальнева А.А., Федотов В.Г. Возбужденные молекулы в реакции фтора с ацетоном (фторацетоны; синтез; механизм). –Кинетика и катализ, 1981, т.22, в.5, с.1095-1099.
136. Капралова Г.А., Марголина Е.М., Русин Л.Ю., Чайкин А.М., Шилов А.Е. Роль колебательно-возбужденных молекул в реакциях фторирования молекулярным фтором. –В сб. «5 Междунар. симпозиум по химии фтора», Тезисы докл., М., : Наука, 1969, с.102-104.
137. Кнунянц И.Л, Гамбарян Н.П., Рохлин Е.М. Карбены. (Соединения двухвалентного углерода, промежуточно образующиеся в органических реакциях). –Успехи химии, 1958, т.27, в.12, с.1361-1436.
138. Мухаметшин Ф.М., Жирнов О.М., Айнагос Н.А., Петров Ю.И., Ляпунов М.И. О некоторых особенностях гипофторитного метода получения перфторметилвинилового эфира. Сообщ. 2. Синтез трифторметилгипофторита. –Тр. ГИПХ, № 69, инв. № Т-2769, Л., 1980, с.73-78.
139. Мухаметшин Ф.М., Поврозник С.В., Жирнов О.М. Исследование кинетики и механизма каталитической реакции образования трифторметилгипофторита. –Тр. ГИПХ, № 103, инв. № Т-2981, Л., 1983, с.75-80.
140. Авторское свидетельство СССР 1141707. Способ получения 1,1-дифтор-1-хлорэтана или 1,1,2,2-тетрафтордихлорэтана. /Захаров В.Ю., Голдинов А.Л., Боровнев Л. М., Голубев А.Н., Калашникова Н.А., Захарова О.М., Любимова Л.А.
141. Капралова Г.А., Бубен С.Н., Чайкин А.М. Кинетика и механизм реакции фторирования окиси углерода. –Кинетика и катализ, 1975, т.16, в.3, с.591-595.
142. Поврозник С.В. Разработка способа и технологии получения фторокситрифторметана реакциями элементарного фтора с карбонилсодержащими соединениями. Дисс. канд. техн. наук, 1987, ПФ НПО ГИПХ, 161с.
143. Краснов К.С. Молекулы и химическая связь. –М.: Высш. школа, 1977, с.117.
144. Безмелицын В.Н., Легасов В.А., Чайванов Б.Б. Синтез дифторида криптона с применением термической генерации потоков атомарного фтора. –Докл. АН СССР. Сер. хим., 1977, т.235, № 1, с.96-98.
145. Безмелицын В.Н., Легасов В.А., Спирин С.Н., Чайванов Б.Б. Кинетика каталитической атомизации фтора. –Докл. АН СССР. Сер.физ.химия, 1982, т.262, № 5, с.1153-1157.
146. Палкина Л.А., Спирин С.Н., Тищенко П.П. процессы с участием атомарного фтора. –Докл. АН СССР. Сер.физ.химия, 1982, т.262, с.1428-1433.
147. Безмелицын В.Н., Васильев А.А., Синянский В.Ф., Чайванов Б.Б. Кинетика термокаталитической диссоциации фтора на поверхности никеля. –
Тезисы докладов на 8 Всесоюзн. симп. по химии неорг. фторидов, 1987, М.: Наука, с.59.
148. Никоноров Ю.И. Фториды никеля. Их каталитическая и окислительная активность. –Тезисы докладов на 5 Всесоюзн. симп. по химии неорг. фторидов, 1978, М.: Наука, с.205.
149. Рысс И.Г. Химия фтора и его неорганических соединений. –М.: Госхимиздат, 1956, с.576-579.
150. Redwood M.E., Willis C. J. Fully fluorinated alkoxides. PartІ. Trifluoromethoxides of alkali metals. –Canad. J.Chem., 1965,v.43,p.1893-1898.
151. Губанов В.А., Веретенников Н.В., Тройчанская П.Е., Долгопольский И.М. Синтез перфторалкоксидов металлов І группы и термографическое изучение их свойств. –Ж. Орг. хим., 1975, т.ІІ, № 2, с.322-325.
152. Levy J. B., Kennedy R. C. Bis (trifluoromethyl) peroxide.І. Thermodynamics of the equilibrium with carbonyl fluoride and trifluoromethyl hypofluorite. –J. Am. Chem. Soc., 1972, v.94, no 10, p.3302-3305.
153. Патент Японии 35888. Способ получения трифторметилгипофторита. / Нагасэ Сюндзи, Абэ Такаси, Баба Хадзимэ, Охира Кадзуо. –Опубл. 09.09.72, РЖХимия, 1973, 14Н120.
154. Патент США 3687825. Способ получения трифторметилгипофторита. Нагасэ Сюндзи, Абэ Такаси, Баба Надзимэ. –Опубл. 29.08.72, РЖ Химия, 1973, 21Н26.
155. Патент США 2983764. Способ получения фторуглеродных соединений./ Кнак Д.Ф. –Опубл. 9.05.61, РЖ Химия, 1962, 14Л53.
156. Патент Японии 56-16429. Олигомеризация гексафторпропилена в газовой фазе. / Хосака Йоносукэ, Хигасидзука Такэси. –Опубл. 17.02.81, РЖ Химия, 1982, 3Н10.
157. Патент Японии 50-45818. Получение олигомеров гексафторпропилена. Кадзава Масахиро, Комацу Тадааки, Мацуока Кимиаки. –Опубл. 08.06.81, РЖ Химия, 1982, 9НІІ.
158. Патент США 4296265. Способ получения олигомеров гексафторпропилена. / Хосака Йоносукэ, Тозука Такаси. –Опубл. 20.10.81, РЖ Химия, 1982, І3НІІ.
159. Патент Германии 3027229. Способ получения олигомеров гексафторпропилена. / Осака Ю., Тозука Т. –Опубл. 07.02.81, Изобретения в СССР и за рубежом, 1981, В.55, № 12, с.30.
160. Патент Бельгии 2055824А. Способ получения олигомеров гексафторпропилена. /Осака И., Тозука Т. –Опубл. 18.07.80, Сборник рефератов НИОКР, обзоров, переводов и деп. рукописей. ДСП. Химия и химическая технология, М., 1986, № 5, с.14.
161. Патент Германии 2616733. Способ получения ди
-и тримеров гексафторпропилена. / Окава М., Комацу Т., Мацуока К. –Опубл. 20.03.80, Изобретения в СССР и за рубежом, 1980, № 15, с.13.
162. Патент Японии 57-2697. Способ получения олигомеров гексафторпропена. / Нэосу К.К. –Опубл. 18.01.82, Изобретения в СССР и за рубежом, 1982, в.57, № 14, с.113.
163. Scherer K. V., Ono T., Jamanouchi K., FernandezR., Henderson P.B. F-2,4-Dimethyl-3-ethyl-3-pentyl and F-2,4-dimethyl-3-isopropyl-3pentyl: stable tert-perfluoroalkyl radicals prepared by addition of fluorine or trifluoromethyl to a perfluoroalkene. –J. Am. Chem. Soc., 1985, v.107, № 3, p.718-719.
164. Scherer K. V., Fernandez R., Henderson P.B. Long lived perfluoroalkyl radicals: preparation by direct fluorination and alternate routes, properties and rates andmechanisms of disappearance. –Int. Symp. “Centenary of the discovery of fluorine”, Abstr., Paris, 1986, p.1.
165. Патент Германии 2332097. Способ получения перфторнонанов. /Халац С.П. –Опубл.16.01. 75,РЖ Химия, 1975, 18Н25.
166. Патент Германии 2332088. Способ получения перфтор-2-метилпентана./ Халац С.П. –Опубл. 16.07.81, Изобретения в СССР и зарубежом, 1981, в.55, № 23, с.21.
167. Аллаяров С.Р., Баркалов И.М., Гольданский В.И., Кирюхин Д.П. Образование стабильных радикалов из фторорганических соединений. –Изв. АН СССР. Сер. хим., 1983, No 6, с.1225-1228.
168. Аллаяров С.Р., Баркалов И.М., Гольданский В.И., Демидов С.В., Кирюхин Д.П., Михайлов А.И. Стабильные радикалы –растворенные перфторалкилы. –Изв. АН СССР. Сер. хим., 1983, № 6, с.1448-1449.
169. Аллаяров С.Р., Михайлов А.И., Баркалов И.М. Новый стабильный перфторалкильный радикал. –Изв. АН СССР. Сер.хим., 1985, № 7, с.1667-1669.
170. Аллаяров С.Р., Баркалов И.М., Лебедов М.Ю., Логинова Н.Н., Михайлов А.И. Образование стабильных радикалов при радиолизе гексафторпропилена. –Изв. АН СССР. Сер. хим., 1986, № 2, с.462-463.
171. Аллаяров С.Р., Баркалов И.М., Гольданский В.И. Образование долгоживущих радикалов при фторировании жидких углеводородов. – Изв. АН СССР. Сер. хим., 1986, № 10, с.394.
172. Аллаяров С.Р., Сумина И.В., Баркалов И.М., Михайлов А.И. Образование стабильных радикалов при
фотолизе перфтор-2,4-диметил-3-этилпентена-2. –Изв. АН СССР, Сер. хим., 1986, № 10, с.2359-2361.
173. Krusic P. J., Scherer K. V. An electron spin resonance study of long lived fluoroalkyl radicals generated by photolysis of hindered perfluoroalkenes. Int. Symp. “Centenary of the discovery of fluorine”. Abstr., Paris, 1986, p.37.
174. Levy J.B., Kennedy R. C. Homolytic displacement reactions of carbon.І. The fluorine-perfluorocyclobutane reactions. –J.Am.Chem.Soc., 1974, v.96, № 15, p.4791-9795.
175. Гольдинов А.Л., Захаров В.Ю., Байбаков П.Я., Жукова В.А., Новикова М.Д. Новые аспекты прямого фторирования. Каталитическое газофазное фторирование фторолефинов; получение перфторуглеродов. –Отчет предприятия п/я А-1619, 1987г., инв. № 1288, 87с.
176. Матвеев С.А., Прокудин Л.П., Тимофеев Н.В. Использование трифторида кобальта для фторирования органических соединений по двойным связям. –Тр. ГИПХ, 1975, № 31, инв. № Т-1728, с.119-127.
177. Асович В.С., Прокудин И.П. Фторирование ацетилена фторидами кобальта и церия. Тр. ГИПХ, № 55, инв. № Т-2552, Л., 1978, с.21-24.
178. Асович В.С., Прокудин И.П., Максимов Б.Н., Степанов В.П. Изучение кинетических закономерностей фторирования ацетилена трехфтористым кобальтом. –Тр. ГИПХ, № 65, инв. № Т-2723, Л., 1979, с.42-47.
179. Котиков С.В., Прокудин И.П., Асович В.С. Изучение кинетических закономерностей фторирования гексафторпропилена трехфтористым кобальтом. –Тр. ГИПХ, № 101,инв. №Т-2986,Л.,ГИПХ, 1985,с.24-27.
180. Burdon J., Knights J.R., Parsons J. W., Tatlow J. C. The fluorination of ethane and ethene over potassium tetrafluorocobaltate (ІІІ) and cobalt trifluoride. –Tetrahedron, 1976, v.32, p.1041-1043.
181. Гольдинов А.Л., Боровнев Л.М., Широкова Н.С., Ковбасюк О.Г., Тишина В.В. Влияние технологических параметров синтеза и чистоты растворителя на свойства фторопласта-50. –Отчет предприятия п/я А-1619, 1987, инв. № 1231.
182. Патент Германии 2451493. Способ получения перфторированных эфиров./ Халац С.П., Клаг Ф. –Опубл.06.05.76,ИзобретениявСССРизарубежом,1982,в.57, № 22,с.5.
183. Patent US 3665041 (USA). Perfluorinated polyethers and process for their preparation. / Sianesi D., Fontanelli R., -Publ. 23.05.72.
184. Рябинин Н.А., Долубенов А.Е., Шматов Л.Е. Фторполиэфиры как антиадгезивы при прессовании перхлората нитрония. –Тр. ГИПХ, № 101, инв. № Т-2986, Л., 1985, с.83-87.
185. Авторское свидетельство СССР 1332758. Способ очистки гексафторпропилена, тетрафторэтилена и их семей от октафторизобутилена. /Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
186. Гольдинов А.Л., Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Разработка технологии получения 2
-перфторметил-4-оксаперфторнонана на основе перфторизобутилена и 1,1,5-тригидроперфтор пентанола. –Отчет предприятия п/я А-1619, 1987, инв. № 1229, 39с.
187. Авторское свидетельство СССР 1148285. 3-Гидропропилфторсульфат в качестве промежуточного продукта для синтеза 2,2,3,3-тетрафторпропионовой кислоты и способ ее получения. /Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
188. Заявка СССР 3966753. Способ получения 2-гидротетрафторэтилфторсульфата. /Фокин А.В., Рапкин А.И., Татаринов А.С. и др. Пол. реш. от 26.09.86.
189. Авторское свидетельство СССР 1233595. Способ получения ω-гидроперфторкарбоновых кислот (его варианты). / Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
190. Технические требования к ПФДМЦГ для использования в радиоэлектронике. М.: ИНЭОС АН СССР. Рег. № 12111-10 ДСП от 18.01.85.
191. Исходные данные для разработки жидкости-диэлектрика. М.: ИНЭОС АН СССР. Прил. к вх. № 155 от 18.02.85.
192. Гольдинов А.Л., Захаров В.Ю., Байбаков П.Я. и др. Разработка технологии получения перфтордиметилциклогексана повышенной чистоты. –Отчет предприятия п/я А-1619, 1986, инв. № 1218, 24с.
193. Гольдинов А.Л., Масляков А.И., Захаров В.Ю. и др. О возможности резкого увеличения выпуска дефицитных фторуглеродных жидкостей и смазок. –Отчет предприятия п/я А-1619, 1986, № 1219, 13с.
194. Временный технологический регламент производства перфтордекалина.1981,инв. № 1115.
195. Furin G.G. Synthetic aspects of the fluorination of organic compounds. (Ed.) M.E. Vol’pin./Harwood Academic
Publishers GmbH: United Kingdom, Sov. Sci. Rev., B. Chem., 1991, V.16, Part 1,р.140.
196. Rozen S., Kol M. Olefin epoxidation using elemental fluorine. / J.Org.Chem., 1990. V.55,р.5155-5159.
197. Patent JP 02-131438, (1990). CO719/08. Preparation of hexafluoroethane. /Shimuzu M.C.A. 1990, V.I 13,97031.
198.PatentJP62-193946, (1989).CO719/08,CO17/04. Способ получения фторсодержащих этанов. /Фарука Я., Коно Х. РЖХ, 1990, 2Н 131.
199. Lagow R.J. Important new developments in direct fluorination technology. /Memorial symposium for Prof. Nobuo Ishikawa, Dec. 9-10, 1991, Nippon Kaiun Club, Japan.
200. Lin W.H., Lagow R.J. Synthesis of perfluorodicyclohexano-crown-6 ether. /J.Chem.Soc., Chem.Commun., 1991,р.13-14.
201. Clark W.D., Lin T.Y., Maleknia S.D., Lagow R.J. Synthesis of perfluorotetralkyl ortocarbonates using elemental fluorine. / J.Org.Chem., 1989. V.54.р.1990-1992.
202. Sievert A.C., Tong W.R., Nappa M.J. Preparation D’analogues fluores de la pheromone d’anthanomus grandis. / J.Fluorine Chem., 1991. V.51.р.397-405.
203. Tonelli C., Tortelli V. Photoinduced fluorination of hexafluoropropene trimers: synthesis of branched perfluoroalkanes. /J.Chem.Soc., Perkin Trans I, 1990, № 1,р.23-26.
204. Patent Application EP 344935, (1989). CO8G65/00. Preparation of prefluoroethers by photo-assisted fluorination. / Nappa M.J., Sievert A. C.A. 1990, V. 112, 216229.
205. Patent US 4960951, (1990). CO743/12. Novel perfluoroalkyl ethers by two-step fluorination of polyols./ Nappa M.J., Sievert A.C. Tong W.R. C.A. 1991, V. 111, 216229.
206. Sievert A.C., Tong W.R., Nappa M.J. Synthesis of perfluorinated ethers by an improved solution phase direct fluorination process. / J.Fluorine Chem. 1991.V. 53, р. 397-417.
207. Мизин Г.Г., Симаков Б.В., Шкультецкая Л.В., Молдавский Д.Д. Исследование способа получения перфторэтилциклогексена. / ЖПХ. 1991. т.64 ,в.6. с.293-1296.
208. Moldavsky D.D., Mizin G.G., Shultezkaya L.V., Simakov B.V., Furin G.G. Synthesis of perfluoroethylcyclohexene, its alkoxy and diakylamino derivates and their electrochemical fluorination. / Abstracts of 10-European Symposium on Fluorine Chemistry, Sept. 20-25, 1992, Padua, Italy. Abstracts.B7;J.FluorineChem., 1992.v.58. р.185.
209. Молдавский Д. Д. Перфторорганические соединения: Получение прямым фторированиемуглеводородов. Дисс. докт. хим. наук, 2002, РНЦ Прикладная химия, 268с.
210. Суворов Б.В., Букейханов Н.Р. Окислительные реакции в органическом синтезе. 1978г. М., Химия, 197с.
211. Семенова Г.А., Лейтес И.Л., Аксельрод Ю.В., Маркина М.И., Сергеев С.П.,Харьковская Е.Н. Каталитические и адсорбционные методы очистки газов от сернистых соединений. –Очистка технологических газов. 1977г, М., Химия, 1977, с. 35-37.
212. Панов Г.И. Закономерности гетерогенно-каталитической активации двухатомных молекул (N2, О2, Н2). Новые каталитические системы активации молекулярного азота. Дисс. докт. хим. наук, 1986г. Новосибирск, 374с.
213. Авторское свидетельство ЧССР 220740. Способ получения тетрахлортетрафторпропана. –Паллета О., П. Свобода, Л. Стефан, Я. Квисала. –Опубл. 15.12.85. РЖ Химия, 1986, ІІН24.
214. Патент Японии 60-116637. Получение фторметана. –Такаяма С., Мэйраку Ф., Такоити А., Ковасаки Х. –Опубл. 24.06.85. РЖ Химия, 1986, 9Н16.
215. Марголис Л.Я. Окисление углеводородов на гетерогенных катализаторах. 1977г. М., Химия, с. 296.
216. Каваев М.М., Запорин А.П., Клещев Н.Ф. Каталитическое окисление аммиака. 1983г. М., Химия, с. 31.
217. Технология катализаторов. 1974г. Л.,Химия,с. 138-140.
218. Patent US 2760997. Process for the production of chlorotrifluoroethylene by passing a mixture of trichlorotrifluoroethane and hydrogen through an unobstructed iron tube. / Rucker J. T., Stormon D. B. –Publ. 28.08.56.
219. Patent US 2615925. Preparation of olefinic compounds. / Bordner C.A. –Publ. 28.10.52.
220. Patent US 2704777. Preparation of halogenated olefines / Clark J.W. –Publ. 22.03.55.
221. Patent GB 698386. Improvement in preparation of halogenated olefines / Clark J. W. –Publ. 14.10.53.
222. Patent DE 1044800. Verfahren zur Herstellung von Chlortrifluorathylen / Clark J. W. –Publ. 27.11.58.
223. Patent US 2685606. Preparation of halogenated olefines / Clark J. W. –Publ. 03.08.54 C.A. 48, 127881 (1954).
224. Patent US 2697124. Dehalogenation of fluorohalocarbons / Russel M.M. –Publ. 14.12.54 C. A. 50, 2650 (1956).
225. Patent US 2864873. Production of 1,1,2-trifluoro-2-chloroethylene / Miller C. B., Smith L. B. –Publ. 16.12.58.
226. Патент США 3043889. Получение фтористого этилена. / Смит Л.Б., Вулф К. –Опубл. 10.07.62. РЖ Химия, 1964, 3Н24.
227. Патент Германии 1186847. Получение 1-хлор-1,2,2-трифторэтилена. / Смит Л.Б., Вульф К. –Опубл. 4.10.65, РЖ Химия, 1966, 13Н26.
228. Патент США 3333011. Получение хлортрифторэтилена./Анелло Л.Г., Вульф К. –Опубл. 25.07.67 РЖ Химия, 1969, 14Н30.
229. Патент Японии 60-185734. Получение трифторхлорэтилена. / Моримото Такэси, Морикава Санэсукэ, Фунаяма Кэйди. –Опубл.21.09.85, 19Н37.
230. Европейский патент 0053657. Способ получения хлортрифторэтилена и трифторэтилена. / Канинхэм У.Д., Плэкорз Р.Е., Смит Э.М. –Опубл. 16.06.82. Изобр. в СССР и з/р., вып. 57, № 15, с. 83.
231. Патент Японии 43-8454. Способ получения трифторэтилена. / Саката Хиросукэ. –Опубл. 02.04.68. РЖХимия, 1969, 3Н36.
232. Патент США 2913400. Катализатор из окиси молибдена для конверсии углеводородов. / Буртон У.П., Лефрэнкойс Ф.А., Риблет Е.У. –Опубл. 17.11.59, РЖХимия, 1961, 9К156.
233. Патент США 3354233. Производство тетрафторэтилена. / Анелло Л.Г., Вульф К. –Опубл. 21.11.67, РЖ Химия, 1969, 9Н32.
234. Patent DE 1270547. Verfahren zur Herstellung von Tetrafluorathylen/ Anello Z. G., Wolf C. –Publ. 30.01.69.
235. Патент США 3505417. Дегалоидирование фторгалогенуглеродов. / Гарднер Л.Е. –Опубл. 07.04.70, РЖ Химия, 1971, 11133.
236. Патент США 3636173. Способ каталитического дегалоидирования./ Гарднер Л.Е. –Опубл. 18.01.72, РЖ Химия, 1972, 21Н21.
237. Патент США 3789016. Катализатор гидродегалоидирования. / Гарднер Л.Е. –Опубл. 29.01.74. РЖ Химия, 1975, 2Н217.
238. Патент США 3636172. Дегалоидирование фторгалоидуглеводородов. / Гарднер Л.Е. –Опубл. 18.01.72, РЖ Химия, 1972, 21Н20.
239. Патент США 2900423. Производство перфторпропилена. / Смит Л.Б. –Опубл. 18.08.59. РЖ Химия, 1961, 7Л44.
240. Патент США 2734090. Получение фтористого винилидена. / Кафи Д.Д., Миллер Ч.Б. –Опубл. 07.02.56, РЖ Химия, 1957, 12, 42313.
241. Патент США 3404180. Получение арбонилфторида. / К.К. Лестер. –Опубл. 1.10.68. –РЖ Химия, 1969, 23Н92.
242. Патент Франции 1460246. Способ получения окиси тетрафторэтилена. / Эдисон. –Опубл. 17.10.66. –РЖ Химия, 1968 ІІНІ24.
243. Соколов Л.Ф. Исследование и разработка технологии процессов окисления фторолефинов. –
Дисс. докт. хим. наук. –Ленинград, ВНИИСК, 1979.
244. Авторское свидетельство СССР 190555. Способ получения фтористого водорода. / Гольдинов А.Л., Романов Е.И., Захаров В.Ю. и др.
245. Гольдинов А.Л., Боровнев Л.М., Захаров В.Ю. и др. Термическое обезвреживание фторорганических отходов в водородо-воздушном пламени на опытно-
промышленной установке. –Отчет предприятия п/я А-
1619, 1982, инв. № 887, 18с.
246. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Катализ и инициирование деструктивного окисления, тетрафторэтилена молекулярным
кислородом. Получение карбонилфторида. –Отчет предприятия п/я А-1619, 1983, инв. № 1110, 21с.
247. Патент ГДР 213342. Способ получения фторсодержащих галоидуглеводородов со средней степенью фторирования. / Л. Ингэбург, К. Рэйнфред, М. Дитэр. –Опубл. 24.12.85. –РЖ Химия, 1986, 21Н17.
248. Патент ГДР 203314. Катализатор получения хлорфторметана в газовой фазе. / И. Лелид, Р. Каден, Д. Мросс. –Опубл. 19.10.83. –РЖ Химия, 1984, 17Н181.
249. Патент США 4504686. Способ получения 1,1,2-трифтор-2-хлорэтилдифторметилового эфира. / С. Таками. –Опубл. 12.03.85. –РЖ Химия, 1986, 8Н9.
250. Патент Японии 55-85564. Получение β-трифторметилхлорпиридинов./ Р.Нишима, К. Рудзикама, И.Йокомити, Я. Цудзии, С. Нисимура. –Опубл. 27.06.80. –РЖ Химия, 1981, 19Н177.
251. Патент США 4139568. Способ получения метилфторида. / В. Даниэль, М. Либбс. –Опубл. 13.02.79. –РЖ Химия, 1979, 18Н18.
252. Патент США 4145368. Получение 1,1,1-трифтор-2,2-дихлорэтана. Р. Свинэй, С. Бернард. –Опубл. 20.03.79. –РЖ Химия, 1979, 20Н20.
253. Патент Франции 1453510. Способ очистки дихлортетрафторэтана. / Корп. Электрохимия. –Опубл. 16.08.66. –РЖ Химия, 1968, 11Н39.
254. Виленчик Я.М., Якурнова Г.И., Остапенко Э.А. Синтез трифторацетилфторида на основе окиси тетрафторэтилена. –Тр. ГИПХ, 1985, № 101, с. 147-149.
255. Патент Японии 58-35978. Способ получения пентафторпропионилфторида. / А. Йонотаси, А. Йосио. –Опубл. 08.05.83.-Изобретения в СССР и за рубежом, 1984, вып. 57, № 8, ч.2, с. 130.
256. Патент Японии 58-38231. Получение пентафторпропионилфторида./ И. Йоносукэ, А. Йосио, С. Хироюки. –Опубл. 05.03.83.-РЖ Химия, 1984, 5Н46.
257. Патент США 3250808. Фторсодержащие простые эфиры. / Е.Ф. Мур, А. Мильян, Г. Элеутерио. –Опубл. 10.05.66. –РЖ Химия, 1967, 17Н82.
258. Патент США 3250807. Фторсодержащие карбоновые кислоты с эфирными группировками. / Ч.Г. Фритц, Е.Ф. Мур. –Опубл. 10.05.66. –РЖ Химия, 1967, 17Н84.
259. Патент США 3242218. Способ получения фторуглеродных полиэфиров. / В.Миллер. –Опубл. 22.03.66. –РЖ Химия, 1967, 12С232.
260. Патент ГДР 78376. Способ полимеризации и сополимеризации окиси гексафторпропилена в присутствии гексафторпропилена. / Г. Лотхар. –Опубл. 12.12.70. –РЖ Химия, 1971, 21 С. 361.
261. Патент США 3125599. Полимеры окисей фторуглеродных соединений. / Д. Ворнелл. –Опубл.17.03.64. –РЖХимия, 1965, 15С. 277.
262. Patent US 3291843. Fluorinated vinilethers. / Fritz C.G., Selman S., -Publ. 13.12.66.C.A. 1967, 66, 37427 h.
263. Патент США 4377717. Способ получения перфтор-2-метилпентена-2. / Л.Анелло, Р. Свинэй. –Опубл. 22.03.83. –РЖ Химия, 1983, 24Н1311.
264. Кнунянц И.Л., Фокин А.В., Чебурков Ю.А. Четвертый международный симпозиум по химии фтора в Истес-
Парк (Колорадо, США) –ЖВХО им. Д.И.Менделеева, т. 13, № 3, с. 311-321.
265. Патент США 3449389. Первичные фторзамещенные алкоголяты. / Д. Ворнелл. –Опубл. 10.06.69. –РЖ Химия, 1970, 18Н39.
266. Патент США 3322826. Полимеризация гексафторпропиленэпоксида./ Е.Ф.Мур –Опубл. 0.06.67. –РЖ Химия, 1968, 18С 296.
267. Патент Японии 57-45132. Получение перфтор-2-метил-3-оксагексаноилфторида. / Я. Масаки, М. Сэйдзи. –Опубл. 13.03.82. –РЖ Химия, 1983, 5Н70.
268. Патент США 3274293. Фторсодержащие простые эфиры. / С.Стенлей. –Опубл. 20.09.66. –РЖ Химия, 1968, 5Н161.
269. Патент Германии 2756919. Способ демеризации окиси гексафторпропилена. / Г. Кюхнэ, Ф. Геллер. –Опубл. 05.07.79. –РЖ Химия, 1980, 14Н22.
270. Патент Германии 2924385. Способ димеризации окиси гексафторпропилена. / Г. Кюхнэ. –Опубл. 07.08.80. –РЖ Химия, 1981, 23Н36.
271. Кинле Х., Бадер Э. Активные угли и их промышленное применение. –Л., Химия, 1984, с. 185-192.
272. Патент США 3321515. Способ получения фторированных карбонильных соединений. / Ф.Мур, С.Мильян. –Опубл. 23.05.67.-РЖ Химия, 1968, 15Н48.
273. Беккер Р.А., Асратян Г.В., Дяткин Б.Л. О роли пространственных факторов в реакциях полифторированных α-окисей с аминами. –ЖОрХ, 1973, т. 9, вып. 8, с. 1644-1648.
274. Кнунянц И.Л., Шокина В.В., Тюленева, В.В., Чебурков Ю.А., Аронов Ю.Е. Производные перфторизомасляной кислоты. –Изв. АНСССР,сер.хим., 1966, № 10,с. 1831-1833.
275. Sianesi D., Passety A., Tarli F., The Chemistry of hexafluoropropene epoxide. –J. Org. Chem., 1966,v. 31, № 7,p. 2312-2316.
276. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Инициирование процесса сополимеризации тетрафторэтилена и гексафторпропилена перфторированными диацильными перекисями, синтезированными на основе окиси гексафторпропилена. –Отчет предприятия п/я А-
1619, 1985, инв. № 1154, 28с.
277. Ашмор. Катализ и инициирование химических реакций. М.: Мир, 1966, с. 62.
278. Патент США 3213134. Дециклизация фторированных циклических эфиров. / Д. Морин. –Опубл. 19.10.65. –РЖ Химия, 1967, 9Н60.
279. Патент США 4302608. Способ изомеризации окиси гексафторпропилена до гексафторацетона. / Э.Скури, Д. Милз. –Опубл. 24.11.81. –РЖ Химия, 1982, 20Н56.
280. Патент Японии 58-62131. Получение гексафторацетона. / О. Йосио, А. Йосимаса, М. Макисукэ. –Опубл. 13.04.83. –РЖ Химия, 1984, 12Н30.
281. Патент Японии 58-62130. Получение гексафторацетона. / А. Йосимаса, М. Макисукэ, С. Юкио. –Опубл. 13.04.83. –РЖ Химия, 1984, 12Н29.
282. Авторское свидетельство СССР 740741. Способ получения гексафторацетона. / Я.М. Виленчик, Г.И. Якурнова, Г.И. Лекомцева, Л.П. Заякина, А.П. Харченко. –Опубл. 15.06.80. –РЖ Химия, 1980, 24Н67.
283. Авторское свидетельство СССР 859348. Способ получения гексафторацетона. / Я.М. Виленчик, Г.И. Якурнова. –Опубл. в Б.И., 1981, № 32, РЖ Химия, 1982, 21Н12.
284. Патент США 4238416. Способ изомеризации фторированных соединений. / Д. Кодуо. –Опубл. 09.12.80. –РЖ Химия, 1981, 17Н65.
285. Киперман С.Л. Введение в кинетику гетерогенных каталитических реакций. М.: Наука, 1964, с. 401.
286. Тюльга Г.М., Губанов В.А., Попова И.С., Петрухно Л.А., Болховец Б.М., Тройчанская П.Е., Долгопольский И.М. Изучение реакции фторангидридов перфторкарбоновых кислот с фторидами щелочных металлов. –ЖОрХ, 1973, т. 14, вып. 11, с. 2339-2346.
287. Коншин А.И., Губанов В.А. Об оценке эффективности способа очистки хладоновых растворов перфторполиоксаметиленацетилфторидов. –Отчет предприятия п/я В-8415.
288. Авторское свидетельство СССР 188404. Фторсополимер винилиденфторида с 3,6,8,10,12-пентаоксаперфтортридеценом для термоморозоагрессивостойких изделий. / Грейс А.М., Ершов А.Е., Захаров В.Ю. и др.
289. Карцов С.В., Валов П.И., Соколов Л.Ф., Соколов С.В. Роль поверхности реакционного сосуда в процессе жидкофазного окисления гексафторпропилена. –Изв. АН СССР, сер. хим., 1978, № 10. с. 2268-2272.
290. Авторское свидетельство 229185. Способ получения перфторполиоксаметиленацетилфторидов. / Захаров В.Ю., Гольдинов А.Л., Боровнев Л.М. и др.
291. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Динамика изменения состава газовой и жидкой фаз в промышленном реакторе синтеза окиси гексафторпропилена в ходе операции окисления. –Отчет предприятия п/я А-1619, 1983, инв. № 1089, 10с.
292. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Динамика образования перфторполиоксаметилен ацетилфторидов в процессе жидкофазного окисления гексафторпропилена кислородом на промышленной установке. –Отчет предприятия п/я А-1619, 1985, инв. № 1165, 22с.
293. Гольдинов А.Л., Боровнев Л.М., Захаров В.Ю. и др. Окисление гексафторпропилена молекулярным кислородом в присутствии 1,2-дибромтетрафторэтана (хладона-114В2). Получение окиси гексафторпропилена. –Отчет предприятия п/я А-1619, 1982, инв. № 1072, 57с.
294. Авторское свидетельство СССР 1128436. Пламягасящая смесь. / Боровнев Л.М., Гусенков М.В., Жукова В.А. и др.
295. Кравченко Н.Н., Попов В.А., Ершов Ю.А., Померанцева Э.Г. Некоторые особенности фотолиза перфторированных полиэфиров. –ВМС, т. 23, № 3, 1981, с. 180-183.
296. Заявка СССР 3011822. Способ регенерации хладона-113. / Паниткова Е.С., Беренблит В.В., Сенюшов А.Н. и др.
297. Зейфман Ю.В., Тер-Габриэлян Е.Г., Ганбарян Н.П., Кнунянц И.Л. Химия перфторизобутилена. –Успехи химии, 1984, т. 53, вып. 3, с. 431-461.
298. Кнунянц И.Л., Шокина В.В., Кулешова А.Д. Присоединение галоидводородов к фторолефинам. –Изв. АН СССР, сер. хим., 1960, № 9, с.1693-1695.
299. Постовой С.А., Мысов Е.И., Зейфман Ю.В., Кнунянц И.Л. Нуклеофильная конденсация октафторизобутилена с тетрафторэтиленом. –Изв. АН СССР, сер. хим., 1982, № 7, с. 1586-1590.
300. Патент США 3389187. Димер перфторизобутилена. / Д.Л. Миллер. –Опубл. 18.06.68. –РЖ Химия, 1969, 17Н3211.
301. Кнунянц И.Л., Герман Л.С., Дяткин Б.П. Реакции фторолефинов. Сообщение ІІ. Взаимодействие соединений ряда перфторизобутилена с аминами и аммиаком. –Изв. АН СССР, отд.хим.н., 1960, № 2, с. 221-230.
302. Патент Японии 53-73504. Удаление октафторизобутилена. / Курода Такэси, Фурукава Йосики, Мацуоха Ну. –Опубл. 30.06.78. РЖХимия, 1979, 10Н2011.
303. Дорогимский В.А., Коломиец А.Ф., Сокольский Г.А. О реакции октафторизобутилена с этиленгликолем и глицерином. –ЖОрХ, 1983, т.19, вып. 8, с.1762-1763.
304. Авторское свидетельство СССР 129653. Способ получения гексафторизомасляной кислоты. / Кнунянц И.Л., Чебурков Ю.А., Красуская М.П. –РЖ Химия, 1961, 9Л75.
305. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Промышленные испытания и внедрение способа очистки газов пиролиза тетрафторэтилена от перфторизобутилена. –Отчет предприятия п/я А-1619, 1981, инв. № 1048, 34с.
306. Гольдинов А.Л., Боровнев Л.М., Захаров В.Ю. и др. Увеличение степени использования сырья в производстве гексафторпропилена путем более полного извлечения октафторциклобутана из кубовой фракции. –Отчет предприятия п/я А-1619, 1982, инв. № 1063, 16с.
307. Авторское свидетельство СССР 180854. Способ получения гексафторпропилена. / Боровнев Л.М., Виноградова А.П., Голубев А.Н. и др.
308. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Комплексная переработка кубовой фракции производства тетрафторэтилена. –Отчет предприятия п/я А-1619, 1985, инв. № 15/107 ДСП, 17с.
309. Авторское свидетельство СССР 1336492. Способ получения 2-хлортетрафторэтилфторсульфата. / Фокин А.В., Рапкин А.И., Татаринов А.С. и др.
310. Авторское свидетельство СССР 1184808. Способ получения монофторсульфата брома. / Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
311. Авторское свидетельство СССР 1239092. Способ получения трис(фторсульфата) брома. / Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
312. Заявка СССР 3982041. 3-(Полифторпропионамидо) пропил-2-оксиэтилдиметиламмоний хлориды, обладающие гипотензивным действием. / Фокин А.В., Ковалев Г.В., Мазанов Л.С. и др.
313. Заявка СССР 3982040. N-Полифторацильные производные γ-амномасляной кислоты, обладающие гипертензивным действием. / Фокин А.В., Ковалев Г.В., Мухина Н.В. и др. Пол. реш. от 26.09.86.
314. Автотское свидетельство СССР 1334655. 3-(ω-Хлорперфторалканоиламидо)пропил-диметил-2-оксиэтиламммоний хлориды в качестве поверхностно-активных веществ. /Фокин А.В., Рапкин А.И., Студнев Ю.Н. и др.
315. Авторское свидетельство СССР 196359. Способ получения ω-хлорперфторкарбоновых кислот и эмульгатор. / Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
316. Патент Германии 3034549. Способ получения перфторированных фторангидридов карбоновых кислот. / Вернер Ш. –Опубл. 29.04.82, –РЖ Химия, 1983, 17Н5711.
317. Промышленные катализаторы и носители. Справочник. –Новосибирск, 1972, с. 246.
318. Т. Ван дер Плас. Текстура и химия поверхности углеродных тел. В кн.: Строение и свойства адсорбентов и катализаторов. –М.: Мир, 1973, с. 436-441.
319. Патрик С. Успехи химии фтора. –М., Л., Химия, 1964, т.1, № 2, с. 336-379.
Материал рекомендован к публикации членом редколлегии С.М. Игумновым
Fluorine Notes, 2014, 95, 1-2


