Поступило в редакцию: май 2014
Fluorine Notes, 2014, 94, 7-8
Синтез 1-йод-3-перфторалкилпропанов и 1-йод-4-перфторалкилбутанов
Bálint Menczinger, Gergely Jakab, Dénes Szabó and József Rábai*
Институт Химии, Университет Eötvös Loránd, п.я.32, H-1518, Будапешт 112, Венгрия
e-mail:
rabai@elte.hu
Аннотация: Предложен простой способ получения перфторалкил-алкил йодидов
Ключевые слова: Фторированные строительные блоки, 3-перфторалкил-пропил йодиды, фосфора (III) йодид.
3-(Перфторалкил)-пропил йодиды (CnF2n+1CH2CH2CH2I, 1a-e, n = 4, 6, 8, 9, 10) являются важными реагентами и строительными блоками для фторхимии [1], материаловедения [2], химии полимеров [3], и разработки лекарств [4].Их синтезы преимущественно основаны на превращении предшествующих F-спиртов в йодиды. Для лабораторного синтеза таких фторсодержащих йодидов вполне допустимо использование реакционных систем: KI/фосфорная кислота [5], NaI/хлортриметилсилан [4] или I2/P(C6H5)3/имидазол [6].
Однако здесь мы показываем, что йодид фосфора (III), получаемый in situ из красного фосфора и йода, реагирует с 3-перфторалкилпропанолами (2a-e) или фтористым бутанолом (2f) с образованием заглавных соединений с высокими выходами при высокой чистоте.

Этот процесс легко можно масштабировать для синтеза нескольких сотен граммов, если регулируемое введение йода в инертной атмосфере осуществляется с использованием «терморасплавной капельной воронки» (Рисунок 1) [7].
Предшествующие 3-перфторалкилпропанолы [8] можно приготовить восстановлением соответствующих аддуктов F-йод-гидрина, полученных присоединением перфторалкилйодидов к аллиловому спирту в присутствии радикальных инициаторов, таких как азо-бис-(изобутиронитрил) [9] или окислительно-восстановительной системы Cu(OAc)2/гидразин гидрат [10]. Упомянутые F-йод-гидрины – это летучие промежуточные соединения, позволяющие приготавливать ценные фтористые строительные блоки, включая также фтористые аллиловые спирты [11].
Полученные продукты охарактеризованы 1H-, 13C и 19F- ЯМР спектрами и ГХ. Последние следы спиртовых примесей можно удалять, обработав их эфирный раствор P2O5/SiO2 (SicaPent) [12], или перегонкой над CaH2 [3].
Эксперимент
Перфторалкил-пропанолы (2a-e) были приготовлены согласно тому, как изложено в [8a], с чистотой по ГХ >98%. Фтористый бутанол 2f был приготовлен аналогично, но с использованием гомоаллилового спирта [13]. 1H-, 13C- и 19F-ЯМР спектры регистрировали с помощью прибора Bruker Avance 250 с использованием 5 мм головки обратного зонда 1H/13C/31P/19F при комнатной температуре. Химические сдвиги (δ) приведены в частях на миллион (ppm) относительно внутреннего стандарта TMS (δ =0.00 для 1H, δ =0.00 для 13C) и относительно CFCl3 в качестве внешнего стандарта (δ =0.00 для 19F). Точки плавления определяли с помощью аппарата Боеция для микро-определения точки плавления и далее не корректировали. Мониторинг реакцийи осуществляли с помощью газовой хроматографии (Hewlett-Packard 5890 Серия II, PONA [сшитая метилсиликоновая смола] в колонке размером 50 м x 0.2мм x 0.5 мм, газ-носитель H2, ПИД детектор; Режим: 120 °C, 5 мин, 10 °C/мин, 250 °C, 5 мин Инж: 250°C, Дет: 280°C).
Типичная процедура (ТП):
1,1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8-Гептадекафтор-11-йод-ундекан (1c)
Высушенный на огне четырехгорлый стеклянный реактор объемом 1 литр был оборудован мешалкой, термометром, ’терморасплавной капельной воронкой’ и конденсатором, присоединенным к аргоновому байпасу. Затем в реактор загрузили перфтороктилпропанол (2c, 574 г, 1.20 молей) и красный фосфор (13.0 г, 0.42 моля P), причем в ’ ’ терморасплавную капельную ворону’ загрузили йод (160 г; 0.63 моля I2). Смесь нагрели на масляной бане до температуры примерно 100°C, пока фтористый спирт не расплавился, затем начали перемешивание, а добавление йода регулировали подачей тока горячего воздуха с помощью термофена; на завершение добавления йода требуется около 60 минут. Реакционную смесь грели и перемешивали в течение 2 часов при температуре внутри смеси 135-140°C. Масляную баню отставили, реакционной смеси дали остыть до ~40°C, а продукт в жидком состоянии отделили от осадка H3PO3 и перенесли в колбу Эрленмейера. После разделения между эфиром (1 л) и водой (0.5 л) и обесцвечивания водным раствором NaHSO3 получен бесцветный экстракт. Его высушили (Na2SO4), профильтровали, а растворитель испарили при дистилляции. Бледно-желтый остаток очистили вакуумной перегонкой и получили 607 г (86%) продукта, бесцветной жидкости с точкой кипения 111°C/16 мм рт.ст., которая отвердевает при комнатной температуре и имеет точку плавления 33-34°C (согласно ГХ: 99.5%).
1H ЯМР (250 MHz, CDCl3) δ 3.24 (t, 2 H, CH2I), 2.08–2.33 (2 m, 4 H, C8F17CH2CH2); 19F ЯМР δ -81.9 (t, 3 F), -114.5 (2 F),
-122.7 (6 F), -123.5 (2 F), -124.2 (2 F), -127.0 (2 F); 13C ЯМР δ 32.30 (t, C8F17CH2); 24.66 (t, CH2CH2I); 3.65 (s, CH2CH2I). Этот продукт дает 1H- 19F-, and 13C ЯМР спектры, точку кипения, точку плавления и чистоту по ГХ такие же, что сообщались ранее нашей лабораторией [12].
1,1,1,2,2,3,3,4,4-Нанофтор-7-йод-гептан (1a)
Rf4-пропанол (2a, 33.4г, 120 ммоль) прореагировали с небольшим избытком PI3 [приготовленного из красного фосфора (1.30 г, 42 ммоль) и йода (16 г, 63 ммоль I2)] в закрытой толстостенной пирекс-трубке объемом 250 мл в течение 3 часов при 135-140°C и получили на выходе 34.0 г (73%) бесцветного масла, точка кипения 70 °C/15 мм рт.ст., (проба ГХ: 98%).
1H ЯМР (250 MHz, CDCl3) δ 3.23 (t, 2 H, CH2I), 2.06–2.32 (2 m, 4 H, C8F17CH2CH2); 19F ЯМР δ -82.3 (t, 3 F), -115.1 (2 F),
-125.5 (2 F), -127.2 (2 F); 13C ЯМР δ 32.1 (t, C8F17CH2); 24.5 (t, CH2CH2I); 3.1 (s, CH2CH2I)
Полученный продукт демонстрировал приемлемые физические свойства и 1H-, 19F-, 13C ЯМР спектры сходные с теми, которые сообщались в [3, 4].
1,1,2,2,3,3,4,5,5,6,6-тридекафтор-9-йод-нонан (1b)
Rf6-пропанол (2b, 227г, 0.60 моля) прореагировали с красным фосфором (6.50 г, 0.21 моля) и йодом (80 г, 0.315 моля I2) согласно Типичной процедуре, в течение 2 часов при 135-140°C, и наработали на выходе 237 г (81%) бесцветного масла, точка кипения = 108°C/20 мм рт.ст. (проба ГХ: 99.5%).
1H ЯМР (250 MHz, CDCl3) δ 3.23 (t, 2 H, CH2I), 2.07–2.34 (2 m, 4 H, C8F17CH2CH2); 19F ЯМР δ -82.1 (t, 3 F), -114.8 (2 F),
-122.8 (2 F), -123.8 (2 F); -124.4 (2F); -127.2(2F). 13C ЯМР δ 32.2 (t, C8F17CH2); 24.6 (t, CH2CH2I); 3.0 (s, CH2CH2I)
Полученный продукт демонстрировал приемлемые физические свойства и ЯМР спектры сходные с теми, которые сообщались в [2, 5].
1,1,1,2,3,3,4,4,5,5,6,6,7,7,8,8-Гексадекафтор-11-йод-2-трифторметил-ундекан (1d)
Изо-Rf9-пропанол (15.84 г, 30 ммоль), красный фосфор (0.325 г, 10.5 ммоль) и йод (4.00г, 15.75 ммоль I2) прореагировали в закрытой толстостенной пирекс-трубке в течение 5 часов при 135-140°C и наработали на выходе: 14.33 г (75%) бесцветного масла, точка кипения = 110°C/1.0 мм рт.ст. (проба ГХ: 98%), точка плавления = 28-29°C.
1H ЯМР (250 MHz, CDCl3) δ 3.25 (t, 2 H, CH2I), 2.07–2.35 (2 m, 4 H, C8F17CH2CH2); 19F ЯМР δ -72.2 - (-72.4) (m, 7 F),
-114.2 (2 F), -115.4 (2 F), -121.2 (2 F); -122.0 (4 F); -123.9 (2F). 13C ЯМР δ 32.3 (t, C8F17CH2); 24.7 (t, CH2CH2I); 4.1 (s, CH2CH2I)
1,1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10-генеикосафтор-13-йод-тридекан (1e)
Rf10-пропанол (2e, 11.56г, 20 ммоль), красный фосфор (0.22g, 7 ммоль) и йод (2.67г, 10.5 ммоль I2) прореагировали в закрытой толстостенной пирекс-трубке в течение 4 часов при 135-140°C и наработали продукт. Сырой продукт (~14 г) перекристаллизовали из циклогексана (90 мл) и получили 11.96 г (87%) белых кристаллов, точка плавления: 75-76 °C (проба ГХ: 97%); согласно литературе [14] точка плавления = 80-82°C.
1H ЯМР (250 MHz, CDCl3) δ 3.25 (t, 2 H, CH2I), 2.08–2.34 (2 m, 4 H, C8F17CH2CH2); 19F ЯМР δ -81.2 (t, 3 F), -114.2 (2 F),
-122.2 (10 F), -123.2 (2 F); -123.9 (2F); -126.6 (2F). 13C ЯМР δ 32.3 (t, C8F17CH2); 24.6 (t, CH2CH2I); 3.8 (s, CH2CH2I)
1,1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8-Гептадекафтор-12-йод-додекан (1f)
Rf8-бутанол (10.1 g, 20 ммоль), красный фосфор (0.22г, 7 ммоль) и йод (2.67 г, 10.5 ммоль I2) прореагировали в закрытой толстостенной пирекс-трубке в течение 9 часов при 135-140°C и наработан продукт. Выход 10.07 г (82%) бесцветной жидкости, после молекулярной перегонки (температура бани: 135-140°C/1.0 мм рт.ст.), точка плавления = 40-42°C (проба ГХ: 97%). Согласно литературе [13] точка плавления = 50-51°C.
1H ЯМР (250 MHz, CDCl3) δ 3.21 (t, 2 H, CH2I), 2.20–1.71 (m, 6 H); 19F ЯМР δ -81.4 (t, 3 F), -114.9 (2 F), -122.5 (6 F),
-123.3 (2 F); -124.4 (2 F); -126.7 (2 F). 13C ЯМР δ 32.9 (s, C8F17CH2); 30.2 (t, one of CH2CH2CH2CF2 ); 21.7 (t, one of CH2CH2CH2CF2 ); 5.2 (s, CH2I).
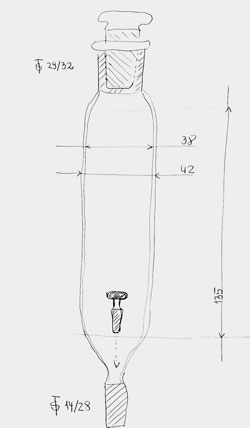
Рисунок 1. Терморасплавная капельная воронка (см. ссылку [7])
‘Терморасплавная’ капельная воронка – это аппарат для пошагового добавления твердых веществ в инертной атмосфере. Он был изготовлен из соединения типа «шип-паз», где паз ST 29/32 и шип ST 14/28, оба имеют длину 60 мм, и стеклянной трубки 38мм длиной 120 мм. Упомянутые соединения привальцованы к стеклянной трубке. Перед использованием аппарата его закрепили в вертикальном положении, и в него поместили небольшой T-образный стеклянный ограничитель (15 мм *6 мм *25 мм), чтобы предотвратить падение твердых частиц, когда их помещают в воронку. ‘Терморасплавная’ капельная воронка может быть заполнена низкоплавкими твердыми веществами, такими как йодгидрин или йод. Затем воронку закрыли с помощью запорного крана ST 29/32 и присоединили к четырехгорлому реактору. Скорость добавления йода легко регулировалась нагреванием нижнего конца воронки ‘горячим воздухом’ из термофена, поскольку T-образный стеклянный ограничитель не препятствует прикапыванию расплавленного йода. Изготовление и применение улучшенной капельной воронки для добавления жидкостей или расплавов уже описано ранее [15].
Список литературы
- (a) Vincent, J.-M.; Rabion, A.; Yachandra, V. K.; Fish, R. H. Angew.
Chem., Int. Ed. Engl. 1997, 36, 2346-2349;
(b) Gladysz, J. A.; Curran, D. P.; Horváth, I. T. (Eds.); Handbook of Fluorous Chemistry Wiley-VCH, Weinheim, (2004). - Tanaka, M.; Rastogi, A.; Toepperwein, G. N.; Riggleman, R. A.; Felix, N. M.; de Pablo, J. J.; Ober,C. K. Chem. Mater. 2009, 21, 3125–3135 3125.
- Sugiyama, K.; Hirao, A.; Nakahama, S.; Macromol. Chem. Phys. 1996, 197, 3149-3165.
- Suzuki, Y.; Kato, T.; Aoyama, H.; Misono, T.; Khlebnikov, V. A.; Mizutani, A.; Ohtake, Y.; Ikeda, T.; Takano, K.; Kwon, H.; Ho, P.-S.; Kim, J.-S.; Lee, W.-I.; Park, C.-H.; Lee, S.-H.; Ahn, S.-O.; (Chugai Seiyaku Kabushika Kaisha, Japan). PCT Int Appl. (2003), WO 2003004515 A1 20030116, 108 pp.
- Brace, N. O.; Marshall, L. W.; Pinson, C. J.; van Wingerden, G. J. Org. Chem. 1984, 49, 2361-2368.
- Kojima, M.; Nakamura, Y.; Ishikawa, T. Tetrahedron Letters 2006, 47, 6309–6314.
- Szijjartó, Cs.; Ivanko, P.; Takács, F. T.; Szabó, D.; Rábai, J.; J. Fluorine Chem. 2008, 129, 386–389.
- (a) Rábai, J.; Szíjjártó, Cs.; Ivanko, P.; Szabó, D. Synthesis
2007, 2581-2584.
(b) JSC "P&M-Invest", Synthesis of Organofluorine Compounds, Ed. Igoumnov S., Igoumnova E., Moscow, 2011, Vol. 2, Chapter 1, pp. 20-29, 31-32. - Brace, N. O.; J. Fluorine Chem. 1999, 93, 1-25.
- Igumnov, S. M.; Don, V. L.; Vyazkov, V. A.; Narinyan, K. E. Mendeleev Commun., 2006, 16 (3), p. 189-190.
- Ivanko, P.; Szíjjártó, Cs.; Szabó, D.; Rábai, J. Fluorine notes, 2012, Vol. 5(84).
- Rábai, J.; Abudurexiti, A.; Szabó, D. In: Gladysz, J. A.; Curran, D. P.; Horvath, I. T. (Eds.) Handbook of Fluorous Chemistry, Weinheim: Wiley - VCH, 2004. pp. 421-423.
- Alvey, L. J.; Meier, R.; Soós, T.; Bernatis, P.; Gladysz, J. A. Eur. J. Inorg. Chem. 2000, 1975-1983.
- Bayardon, J.; Sinou, D. J. Org. Chem. 2004, 69, 3121-3128.
- Mitchell, J., Jr., Henderson, J.W. Anal. Chem. 1950, 22, 374.
Статья рекомендована к публикации членом редколлегии Д.х.н. С.М. Игумновым
Fluorine Notes, 2014, 94, 7-8


