Поступило в редакцию: октябрь 2013
УДК 547.539.546.541.49
Fluorine Notes, 2014, 93, 1-2
РЕАКЦИИ АЛКИЛИРОВАНИЯ, АРИЛИРОВАНИЯ, АЛКЕНИЛИРОВАНИЯ И АЛКИНИЛИРОВАНИЯ ПОЛИФТОРАРЕНОВ И -ГЕТАРЕНОВ, ПРОТЕКАЮЩИЕ ПО АРОМАТИЧЕСКОМУ КОЛЬЦУ
Т.Д.Петрова, В.Е.Платонов
Федеральное государственное бюджетное учреждение науки Новосибирский институт органической химии им.
Н.Н.Ворожцова Сибирского отделения Российской академии наук, 630090 Новосибирск, проспект Акад. Лаврентьева,
9.
e-mail: petrova@nioch.nsc.ru
Аннотация. В обзоре рассмотрены реакции алкилирования, арилирования, алкенилирования и алкинилирования полифтораренов и – гетаренов, протекающие по ароматическому кольцу и приводящие к образованию связей СAr- C. Обзор содержит сведения о традиционных методах проведения таких превращений, которые включают, например, реакции полифтораренов с электрофильными, нуклеофильными, радикальными реагентами. Наряду с этими методами, рассматриваются реакции с применением металлокомплексного катализа. Применение металлокомплексного катализа расширяет область используемых субстратов. Этот метод включает реакции кросс-сочетания металло- и элементоорганических соединений и арилгалогенидов, катализируемые комплексами переходных металлов с различными органическими лигандами. Представлены реакции Кумады, Негиши, Сузуки, Стилла, Хийямы, Хека и Сононогаширы. В ряду полифтораренов и – гетаренов превращения связи CAr-Hal включает также и связь CAr-F.
Ключевые слова: полифторарены и – гетарены, алкилирование, арилирование, алкенилирование, алкинилирование, металлокомплексный катализ, палладиевые катализаторы, реакции кросс-сочетания.
III. Превращение по связи CAr – Hal (Br, Сl, I)
1. Реакции с Na- и Mg-органическими производными
2. Реакции с B-органическими соединениями (реакция Сузуки)
3. Реакции c Cu-органическими соединениями и медный катализ
4. Получение и превращения Zn-органических производных
5. Реакции со Sn- органическими производными (реакция Стилла)
6. Реакции Si-органических производных (реакция Хийямы)
7. Реакции Li-органических соединений
8. Реакции алкенилирования
9. Реакции алкинилирования полифторгалогенаренов (реакция Соногаширы)
В настоящем разделе рассмотрены реакции алкилирования, арилирования (гетарилирования), алкенилирования и алкинилирования полифторгалогеноаренов (Hal = Br, Cl, I), селективно протекающие по связи СAr – Hal с замещением галогена на соответствующую группировку. Большая часть этих реакций основана на прямом нуклеофильном замещении галогена в присутствии различных катализаторов, некоторые включают промежуточное образование полифторированных металлоорганических соединений, которые далее вступают в превращения, приводящие к конечным продуктам.
1. Реакции с Na- и Mg-органическими производными
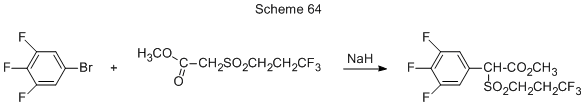
Метил-(3’,4’,5’-трифторфенил-3,3,3-трифторпропилсульфонил)ацетат получается из 3,4,5-трифторбромбензола и метил-(3,3,3-трифторпропилсульфонил)ацетата в присутствии гидрида натрия (схема 64) [158а].
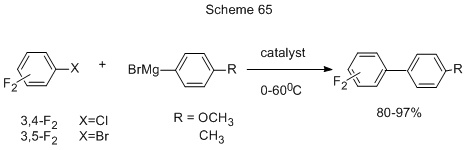
Удобным методом получения несимметричных полифторированных бифенилов является сочетание фенилмагнийбромидов с полифторхлор- или бромбензолами с использованием в качестве эффективного катализатора комбинации N-гетероциклического карбена (NHC) и фторидов Fe, Co, Ni (схема 65) [159].
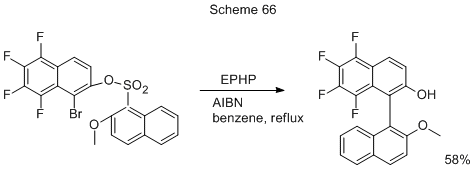
Попытки выйти на метоксипроизводные полифторированных 1,1’-бинафтилов сочетанием 1-бром-2-метокси-5,6,7,8-тетрафторнафталина с 2-метокси-1-нафтилмагнийбромидом или 1-Li-нафтильным производным в присутствии NiCl2(PPh3)2 или, исходя из соответствующего 1-иоднафталинового производного в присутствии Pd(PPh3)4, оказались не слишком успешными из-за низкого выхода (<10%) желаемого бинафтила [40]. Однако подобная задача была успешно решена путём внутримолекулярного радикального кросс-сочетания в полигалогенонафтиловом эфире 2-метоксинафталин-1-сульфоновой кислоты (схема 66) под действием 1-этилпиперидингипофосфита (ЕPHP) и α,α’-азобис-изобутиродинитрила (AIBN) [40]. Предполагается, что идёт радикально-индуцированное 1,5-ипсозамещение через спироциклический интермедиат, способный к реароматизации путём потери SO2.
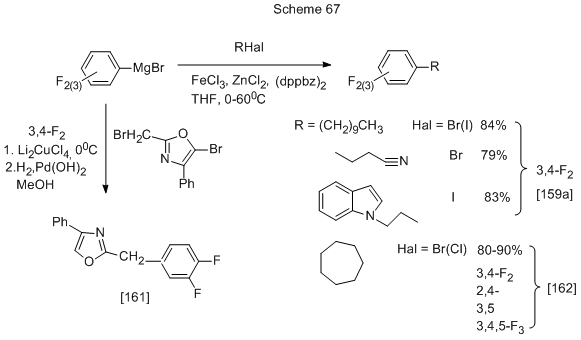
Другой вариант реакции с Mg-органическими производными включает образование Mg-органического соединения с участием галогена исходного полифторфенилгалогеноарена, которое реагирует со своим предшественником или другим галогенопроизводным. Например, при обработке 3,4- и 2,4-дифторбромбензолов магнием в ТГФ и последующем добавлении каталитических количеств (5 мол%) CoCl2 идёт реакция сочетания, и соответствующие тетрафторбифенилы образуются с выходом 48% и 65% [160]. Другие примеры приведены на схеме 67.
Описаны также реакции дифторфенилмагнийбромидов из изомерных дифторбромбензолов (3,4-F2 [163], 3,5-F2 [122, 164, 165], 2,5-F2 [166]), 2,5-дифторфенилмагнийхлорида [167], полифторфенилмагнийбромидов из 2,4,5-трифторбромбензола [168], 2,3,5,6-тетрафтор-4-бромпиридина и бромпентафторбензола [168, 169] с различными карбонильными соединениями, которые идут по карбонильной группе и приводят к соответствующим продуктам оксиалкилирования.
2. Реакции с В-органическими соединениями (реакция Сузуки)
Реакция Сузуки уже рассматривалась в разделе II 2.3. Здесь эта реакция приведена применительно к случаям, когда в исходном полифторгалогеноарене в реакции участвует атом галогена (Br, Cl, I). Примеры с арил- и гетарилборными кислотами даны ниже (схема 68).


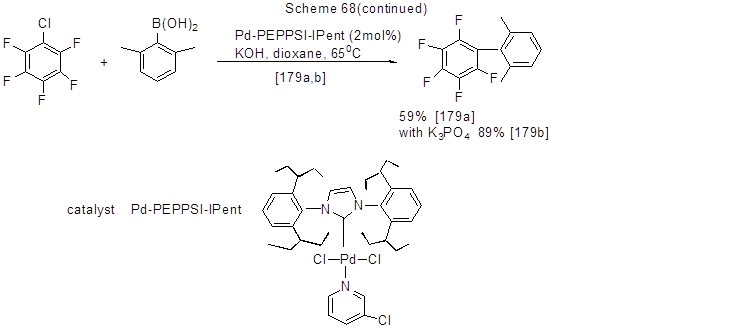
Примерами реакций с фторированными арилборными кислотами служит взаимодействие 3-фторфенил- и 4-фторфенилборных кислот с 2,4-дифториодбензолом в присутствии Na2CO3 и Pd(PPh3)4 , которое приводит к 2,3’,4-трифтор- и 2,4,4’-трифторбифенилам с выходами соответственно 94% и 88% [73].
3. Реакции с Cu-органическими соединениями и медный катализ
Для получения перфторированных разветвлённых олигофениленов, как новых электрон-транспортных материалов для диодов, наиболее важной стадией является реакция кросс-сочетания различных арильных групп, и в этом плане Cu-органохимия даёт наиболее удовлетворительные результаты [180]. На схеме 69 показан путь получения одного из различных типов дендримеров с участием Cu-органических производных.
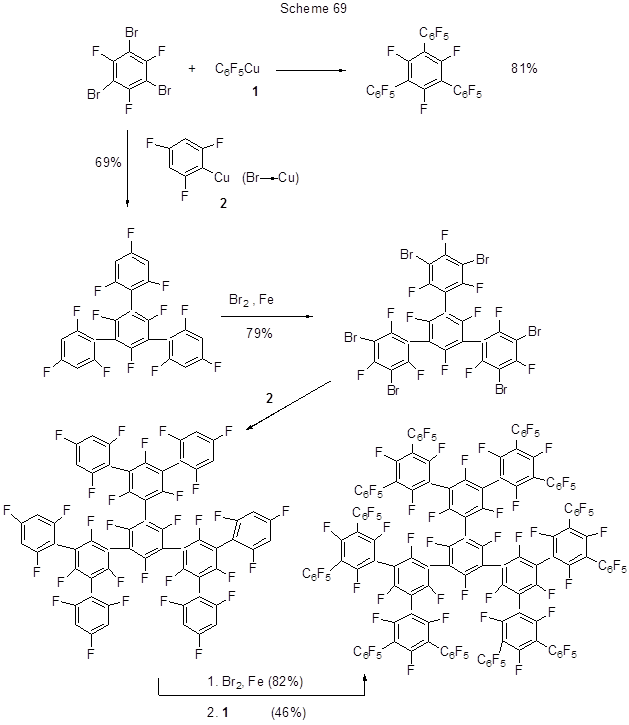
5,5’, 6,6‘, 7,7’, 8,8 ‘- Октафтор-2,2’ –диметокси-1,1’ –бинафтил (1) (схема 70) был получен из 1-бром-2-метокси-5,6,7,8-тетрафторнафталина (2) кросс-сочетанием в присутствии меди по реакции Ульмана с выходом 85% [181]. Попытка использовать этот путь для получения частично фторированных бинафтилов, в частности, 5,6,7,8-тетрафтор-2,2’-диметокси-1,1’-бинафтила (3) кросс-сочетанием исходного бромтетрафторметоксинафталина 2 с 1-бром-2-метоксинафталином в присутствии меди привела к образованию с выходом 85% указанного выше октафтордиметоксибинафтила 1, а ожидаемый тетрафтордиметоксибинафтил 3 получался только с 5%-ным выходом (схема 70) [40].
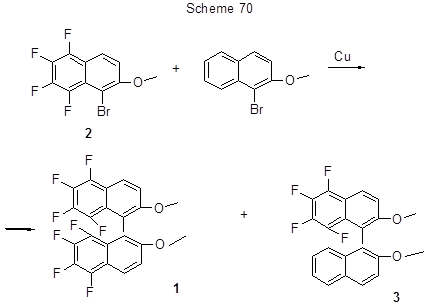
Реакция Ульмана 1-бром-2-метокси-5,6,7,8-тетрафторнафталина 2 с 2-замещёнными 1-бромнафталинами, имеющими в положении 2 акцепторные заместители (COOR, CHO), протекает более успешно. Выходы тетрафторбинафтилов составляют 27-48%. С целью получения 2,2’ –замещённых 5,6,7,8-тетрафтор-1,1’-бинафтилов с более высокими выходами использовали методы окислительного сочетания, которые в отличие от реакции Ульмана более успешны для электронодонорных субстратов. Так, реакция сочетания 4,5,6,7-тетрафтор-2-нафтола и 2-нафтола в присутствии в качестве катализатора 10 мол % Cu(OH)Cl . TМЭДА даёт желаемый тетрафторбинафтил-2,2’-диол с выходом 40%. Реакция дифторбромфенильного производного диметилпиразола с 1-метилимидазолом в присутствии CuI, KI и палладиевого катализатора идёт с замещением брома на гетероциклический остаток (схема 71) [182].
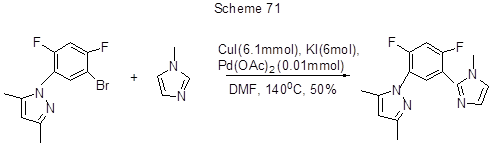
Алкенилирование бромпентафторбензола осуществлено в присутствии кадмия и CuCl (схема 72) [183].
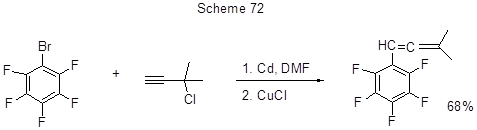
4. Получение и превращения Zn-органических соединений
Аллилполифторарены и –гетарены получаются при взаимодействии Zn-органических реагентов , образующихся из полифторбром- или –хлораренов и гетаренов под действием цинка, с аллилбромидом или аллилхлоридом [184]. Общая схема превращений представлена на схеме 73.
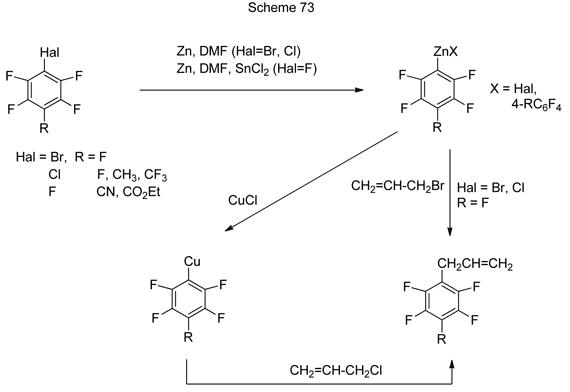
Реакция образующихся из C6F5X (X=Br,Cl) полифторированных Zn-органических реагентов с аллилбромидом идёт путём нуклеофильного замещения атома брома в аллилбромиде на C6F5-группу. Аллильные производные образуются с хорошими выходами и при взаимодействии полифторарилцинкорганических реагентов, образованных из хлорполифтораренов, с аллилхлоридом в присутствии CuCl. Катализ солями меди, по-видимому, обусловлен образованием из Zn-органических реагентов соответствующих Cu-органических соединений, которые далее взаимодействуют с аллилхлоридом. Аналогичным образом протекают превращения 4-хлортетрафторпиридина и 5-хлорнонафториндана при действии на них Zn/DMF и аллилхлорида в присутствии CuCl [184].
Методика получения декафторбифенила высокой чистоты нагреванием бромпентафторбензола с Zn и ацетатом Cu(II) с выходом 70-74% описана в работе [169, C.117-118].
5. Реакции со Sn-органическими производными (реакция Стилла)
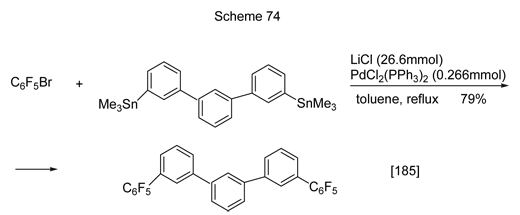
Для осуществления арилирования или гетарилирования бромполифторбензолов использована реакция их кросс-сочетания с олово-аренами или -гетаренами в присутствии палладиевых катализаторов (реакция Стилла). Один из примеров приведён на схеме 74 [185].
Реакция Стилла использована как метод синтеза полифторированных политиофеновых производных – потенциальных полупроводниковых материалов (схема 75) [186].
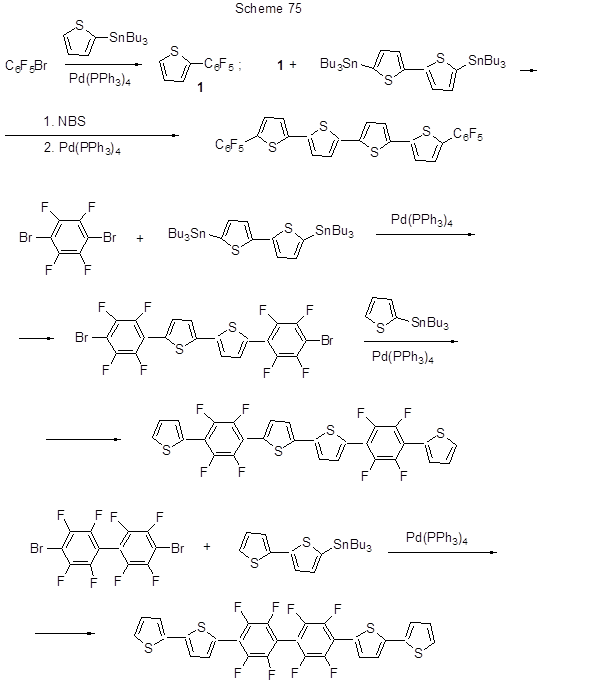
Реакция Sn-производных тиофена с полифторированными иодаренами протекает аналогично (схема 76) [187].
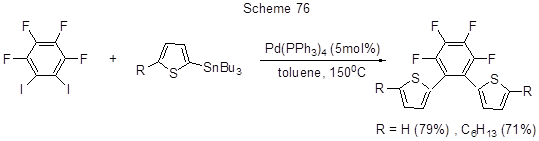
Другой пример полифторарилирования по реакции Стилла как промежуточная стадия в синтезе N-замещённых 5-арилимидазол-2-аминов приведён на схеме 77 [188].
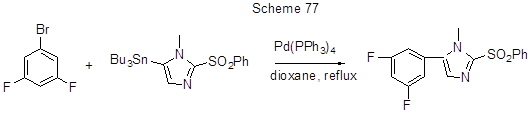
Реакция Стилла 1-бром-2-метокси-5,6,7,8-тетрафторнафталина с (2-метоксинафтил-1)-трибутилоловом не идёт [40]. Реакции ароматических бромдифторпроизводных с трибутил-(пиридин-2-ил) оловом представлены на схеме 78.

Фенилпиридиновые соединения, содержащие электроноакцепторные сульфонильную группу и атомы фтора (левая реакция в схеме 78 [189]), представляют интерес как лиганды для новых иридиевых комплексов, используемых в различных областях. Палладиевый катализатор Pd(AsPh3)4, применяемый в реакции Стилла, образуется in situ из [Pd2 (dba)3] и AsPh3. Другие примеры арилирования пo реакции Стилла, в т.ч. реакция полифторированного Sn-органического производного и нефторированного галогеноарена приведены на схеме 79.
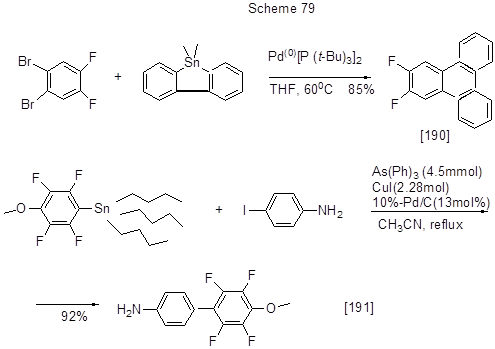
Винильная группа вводится в 6-е положение 5,7-дифтор-6-бромхинолина действием трибутилвинилолова с использованием Pd(PPh3)4 и микроволнового облучения [192] и в 5-е положение 2,3,4-трифтор-5-иод-бензойной кислоты реакцией кислоты с этим реагентом и использованием в качестве катализатора Pd2(dba)3 и 2-фурилфосфина (выход винильного производного 83%) [193].
Возможность использования олово-органического реагента в другого типа реакциях, например, для внутримолекулярного радикального арилирования, продемонстрирована в случае превращения дифторзамещённого С-аллильного аддукта основания Шиффа, полученного из изатина, в орто, орто-дизамещённый дифторбифенил с внутренним 8-членным лактамным кольцом, интересный в плане физиологической активности, которое идёт под действием трибутилгидрида олова в присутствии AlBN (схема 80) [194].

6. Реакции Si-органических производных (реакция Хийямы)
Кросс-сочетание иодпентафторбензола с 2-(триметилсилил)-фураном в присутствии Pd-катализатора и основания по типу реакции Хийямы приводит с выходом 54% к 2-пентафторфенилфурану (схема 81) [195].
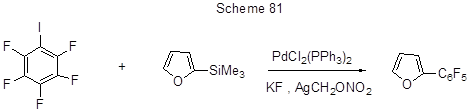
Работа [196, схема 82] иллюстрирует возможность арилирования орто-бромфенолов, в т.ч. и полифторированных, защищённых силильной группой, за счёт того, что эта группировка служит эффективным донором фенильной группы. В ходе внутримолекулярного арилирования, катализируемого Pd-катализатором, предполагается промежуточное образование оксасилоцикла, силильная группа из которого элиминируется, давая бифенильное производное.
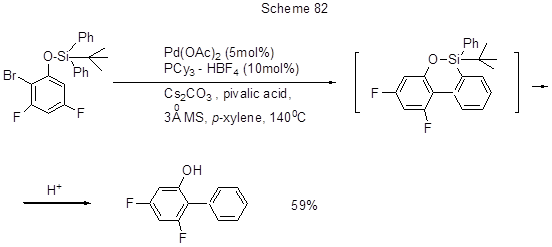
7. Реакции Li-органических соединений
Взаимодействие 1,2-дибром-3,4,5,6-тетрафторбензола с бутиллитием и TiCl4 ведёт к арилированию до 2,2’-дибромоктафторбифенила [197], а из 4-бром-2,6-дифториодбензола с бутиллитием и далее с CuF2 получается 4,4’-дибром- 2,2’,6,6’-тетрафтор-1,1’-бифенил с выходом 79% [67].
Введение вместо атомов брома или иода в полифторарилбромидах или –иодидах оксиалкильной группировки имеет место при взаимодействии соответствующих Li-органических соединений, полученных из этих галогенидов, с карбонильными производными или оксидами. Такое превращение происходит в реакции Li-органических производных из бромпентафторбензола или бромнонафторбифенила с октафторфлуореноном [198], из 1-иод-3,5-дифторбензола - с ацетальдегидом в присутствии тетраметилпиперидина (выход 47%) [80], из 2,3,5-трифторбромбензола, 3,4-дифтор-2-метилбромбензола, 2-метил-3,5-дифториодбензола, 2,3,5,6-тетрафторбромбензола - с пропиленоксидом в присутствии эфирата трёхфтористого бора (выходы оксиалкильных производных соответственно 83%, 71%, 32%, 62%) [199] и из 3,4,5-трифторбромбензола - с циклогексеноксидом также в присутствии эфирата трёхфтористого бора [200].
В некоторых случаях Li-органические соединения, полученные из полифторгалогеноаренов, использовались в реакциях переметаллирования для получения других полифторированных элементоаренов, например, борорганических производных, которые далее вступали в различные превращения. Реакции таких борпроизводных рассмотрены выше в разделе 2.
8. Реакции алкенилирования
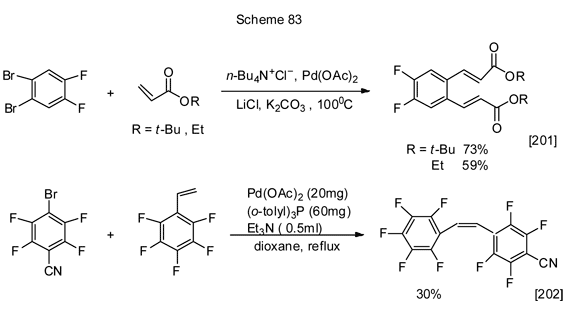
Примеры межмолекулярных реакций получения алкеновых производных полифтораренов из бромполифтораренов по реакции Мизороки-Хека приведены ниже (схема 83).
Реакция может протекать и внутримолекулярно (схема 84).
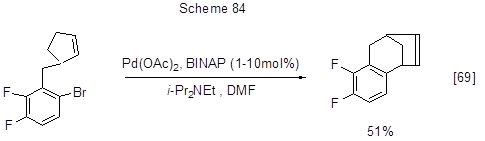
Возможен и внутримолекулярный восстановительный вариант реакции Мизороки-Хека, суть которого состоит в гидроарилировании алкена в α- или β-положение. Так, дигидроинденоизохинолины, в т.ч. дифторсодержащие, интересные в плане их биологической активности, предлагается получать из 3-х компонентной системы (имин , бензоилхлорид, Sn-производное олефина) в две стадии, из которых первая – катализируемое Cu(I) бензоилирование имина до амида и сочетание с олефином, а затем - катализируемое Pd(0) α-арилирование как внутримолекулярная реакция Мизороки-Хека с образованием предполагаемого интермедиата, в котором идёт внутримолекулярное β-арилирование до дигидроинденоизохинолина (схема 85) [203].
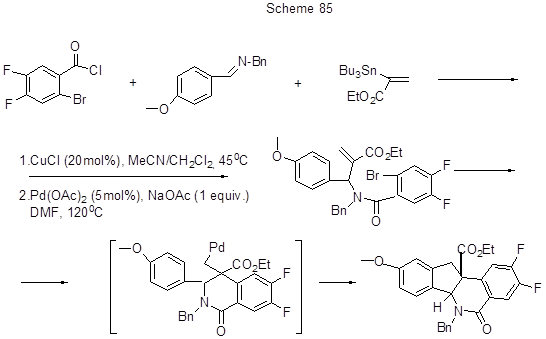
Другая внутримолекулярная реакция (не Мизороки-Хека, но с участием атома брома), позволяющая в конечном итоге получить дифторпроизводное 3,4-дигидро-2(1Н)хинолинона с выходом 70%, предполагает расширение циклопропанового кольца в N-(1’-этокси)циклопропил-2-бром-4,6-дифторанилине (схема 86) [204]. Механизм реакции, по-видимому, включает окислительное присоединение арилбромида к Pd(0) с образованием 4х-членного азапалладиевого цикла, перегруппировку этого цикла с раскрытием циклопропанового кольца и превращение в 7-членный цикл и последующее восстановительное элиминирование с регенерацией Pd(0) и выделением 2-этокси-6,8-дифтор-3,4-дигидрохинoлина. Используемая каталитическая система - Pd2(dba)3 (1.5mol%), дициклогексил-(2’,4’,6’-триспропилбифенил-2-ил)фосфин (7.5mol%) в DMF, основание K2CO3 (1.5equiv.), температура реакции 95°С.

9. Реакции алкинилирования полифторгалогеноаренов (реакция Соногаширы)
Реакция Соногаширы в классическом варианте, т.е. кросс-сочетание галогенопроизводного с алкином в присутствии Pd-катализатора, соли меди и добавкой основания, использована для получения гибридных олигомеров с центральным полифторированным фрагментом, интересных в рамках исследования оптических и электрохимических свойств π-cопряжённых материалов (схема 87, [205]), а также как одна из стадий в синтезе абсорбционных ингибиторов холестерина ряда 3-(арилпропинил)замещённых 2-азетидинонов (схема 88 [206]).

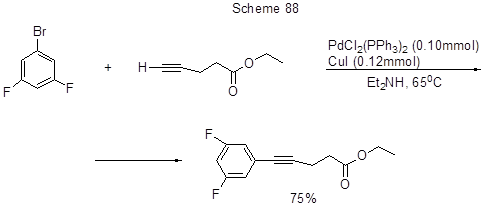
Медный катализ был успешно заменён применением микроволнового облучения (схема 89) [207].
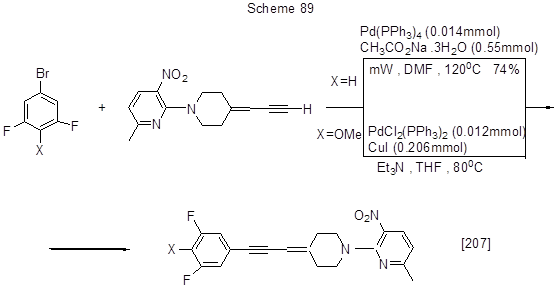
Другие примеры реакции Соногаширы представлены на схеме 90.

О реакциях кросс-сочетания иодпентафторбензола и фенилацетилена в присутствии различных каталитических систем, приводящих к образованию 1-фенил-2-пентафторфенилацетилена, см. [211]. Алкинилирование 3,5-дифторполибромпиридиновых производных по реакции Соногаширы приведено на схеме 91 [212].
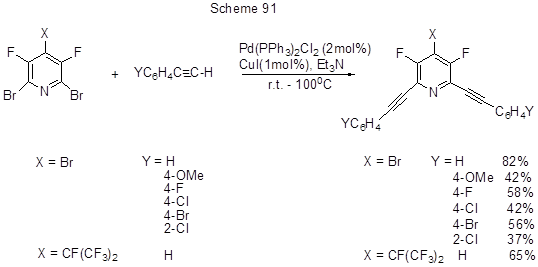
Предполагается, что Pd-катализатор внедряется по более слабой связи CAr-Br, но при этом в реакции участвуют атомы брома в положениях 2,6, т.е. в орто-положениях к атому азота, в отличие от наблюдаемого для нуклеофилов замещения в положение 4. Авторы предполагают, что некоторое влияние на положение замещения может оказывать заряд на атоме азота в переходном состоянии.
IV. Превращения по связи CAr- F
1. Реакции нуклеофильного замещения атома фтора с С-нуклеофилами
1.1 Реакции с Na- органическими соединениями
1.2 Реакции с Li-органическими производными
1.3 Реакции с Mg-органическими производными
1.4 Действие Si-органических производных в присутствии катализаторов, генерирующих фторид-ион.
2. Превращения по связи СAr –F, протекающие с её активацией
Процессы алкилирования, арилирования (гетарилирования) и введения в молекулу полифтораренов группировок с С-С кратными связями реализованы и с участием связи CAr-F. При этом использовались различные методы, прежде всего, наиболее типичные для галогенопроизводных, в т.ч. и полифторированных аренов, реакции нуклеофильного замещения атома фтора, связанного с углеродом ароматического кольца. Наиболее изученными в этом плане являются реакции с С-нуклеофильными реагентами, генерируемыми из различных металл-органических соединений (Li, Na, Mg, Zn, Sn). Однако, в последнее десятилетие в химии органических галогенопроизводных, включая галогеноарены, усиленно развивались новые подходы по вовлечению галогена в превращения путём активации связи СAr –Hal различными способами. Это обусловлено стремлением проводить реакции с более высокими выходами, добиваясь при этом большей региоселективности превращений. Фторарены в этом плане не составили исключения. Недавно появившийся обзор японских авторов «Активация С-F связи в органическом синтезе» [213] суммирует результаты, полученные в этом направлении, и даёт полезный перечень публикаций, связанных с этой проблемой.
1. Реакции нуклеофильного замещения атома фтора с С- нуклеофилами
1.1 Реакции c Na-органическими соединениями
Источник С-анионов – Na-органические соединения – использовался в реакциях нуклеофильного замещения фтора в полифтораренах. Так, при взаимодействии гексафторбензола с Na-производным t-бутилциклопентадиена была получена смесь изомерных пентафторфенил- t-бутилциклопентадиенов и 1,2-бис(пентафторфенил)-4- t-бутил-циклопентадиена (схема 92) [214].
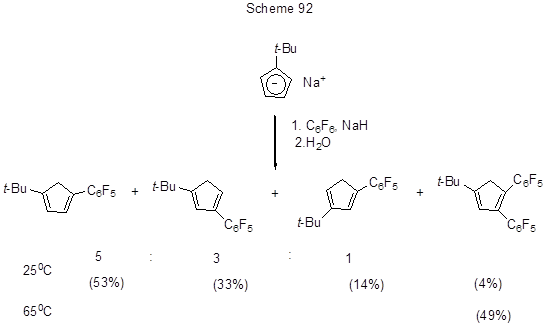
В аналогичной реакции октафтортолуола атаке С-нуклеофила подвергается атом фтора в пара- положении к CF3-группе [215]. 3,4-Дифторнитробензол с малонатом натрия даёт 2-фтор-4-нитрофенилмалонат, который под действием MgCl2 легко претерпевает двойное декарбоксилирование с образованием 2-фтор-4-нитротолуола [216]. 2,4-Дифторнитробензол в этих условиях превращается в 3-фтор-6-нитротолуол (73%). Эта реакция открывает путь получения метилированных нитроароматических соединений.
1.2 Реакции с Li-органическими производными
Традиционными С-нуклеофильными реагентами являются Li-органические соединения. Так, 1-гидрoкси-2-метилпропильная группировка вводится во 2-е положение 4-t-бутокси-2,3,5,6-тетрафторпиридина реакцией этого соединения с Li-CH2CHCH3–CH2OSi(CH3)2t-Bu. Выход продукта 63% [217]. При действии на 1,4-бис(димезитилфосфино)-2,3,5,6-тетрафторбензол н-бутил- и фениллитиевых производных атомы фтора в положениях 2,5 замещаются соответственно на н-бутильную или фенильную группы [218]. Внутримолекулярное превращение реализуется при действии n-BuLi на 2-бром-2’-пентафторфенилбифенил или 2-бром-2’-пентафторфенил-1,1’- бинафтил [219]. При этом с высоким выходом получаются соответственно 1,2,3,4-тетрафтортрифенилен или 7,8,9,10-дибензо-1,2,3,4-тетрафтортрифенилен. В зависимости от количества используемого Li-производного конечные фенилены могут быть получены последовательно или в однореакторном варианте (схема 93).
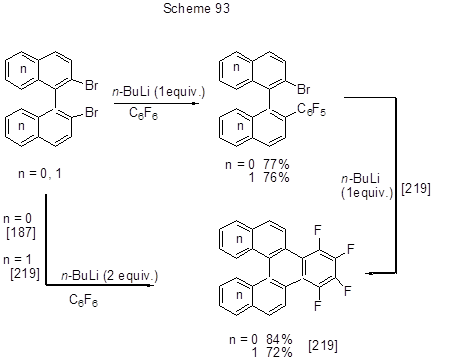
Реакции нуклеофильного замещения 2-пентафторфенил-1-фенилтетрафторэтанов с Li-органическими соединениями протекают только в пара-положение к тетрафторэтильной группе (схема 94) [220].
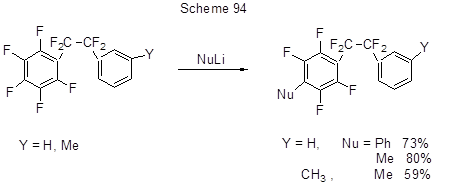
Реакции с Li-органическими производными использовались при синтезе соединений с потенциальными полупроводниковыми свойствами, содержащими в своей структуре полифторированное ароматическое кольцо. Это относится к получению полициклов и, в частности, терфенилов. При этом Li-производные были синтезированы путём обмена H→Li или Br→Li и далее реализовывалось SNAr нуклеофильное замещение фтора в полифторарене (схема 95). Полученные полифторированные терфенилы циклизовали до полифторированных бензо-бис(бензотиофенов), в которых атомы фтора нуклеофильно замещались на этинильные группы при действии Li-производных этилацетилена (схема 95) [221].
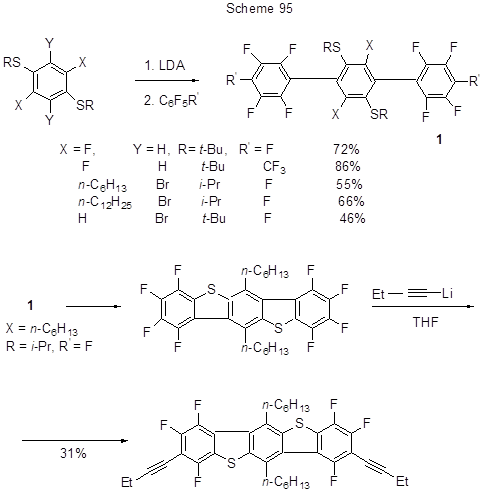
Примеры винилирования в ходе нуклеофильного замещения атома фтора действием Li-этилена см. [40] (схема 96).
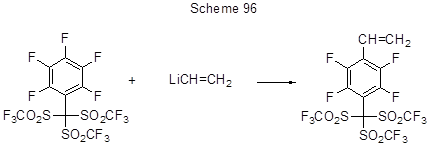
При получении лигандов с полифениленовой структурой использовано замещение пара-атомов фтора в C6F5-фрагментах терфенилов под действием Li-органических производных, а эти фрагменты в терфенилы были введены реакцией гексафторбензола с Li-производными терфенила (схема 97) [185].
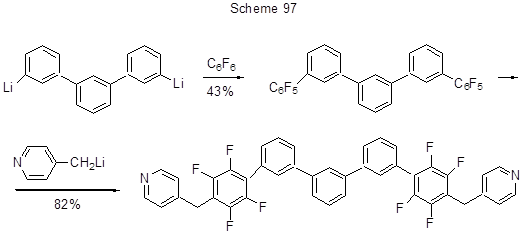
В плане получения полупроводниковых материалов интерес представляют полициклы с различными элементами, внедрёнными в их структуру. На схеме 98 представлен пример получения пентафторфенильного производного 9-оловоантрацена с помощью Li-органического соединения [222].
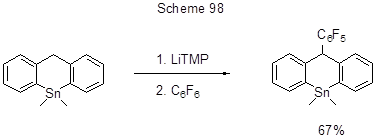
Удобный, с высоким выходом путь синтеза потенциально практически интересных олигофениленов, содержащих перфторированные сегменты, включает, наряду с другими превращениями, сначала реакцию нуклеофильного замещения ароматического атома фтора в декафторбифениле, а затем пара-атомов фтора в образующихся полифениленах под действием Li-производных различных моно-, ди- и полиарилов [223]. На схеме 99 приведён один пример такого превращения при использовании ди-Li-арена.
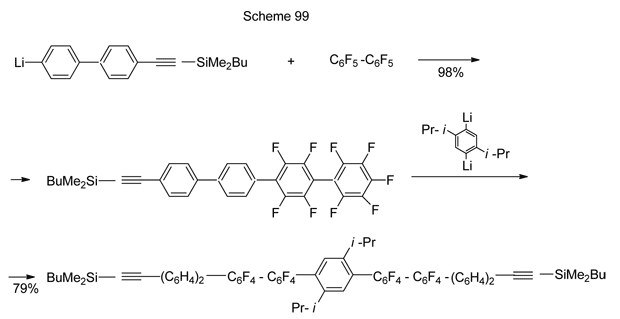
Из реакций нуклеофильного замещения атома фтора в полифтораренах под действием алкил- и ариллитиевых соединений можно указать на синтез 4’-бром-2,3,4,5,6-пентафторбифенила из гексафторбензола и 4-бромфениллития [224], (4-t-бутилтиофенил)-нонафторбифенила из декафторбифенила и 4-t-бутилтиофениллития [225]. пара-Арил- и пара-алкил-тетрафторфенил-бис(трифторметилсульфонил)метаны получаются при замещении пара-атома фтора в пентафторфенил-бис(трифторметилсульфонил)метане действием арил- или алкиллитиевых производных . Продукт реакции, в котором R содержит фенильный радикал с полистирольным остатком в положении 4, обладает высокой кислотностью С-Н связи и представляет собою новую углеродную суперкислоту Брёнстеда, которая проявила себя как эффективный катализатор ацилирования спиртов ангидридами кислот (схема 100) [226].
В работах, связанных с получением полупроводниковых материалов, использованы реакции полифтораренов и с гетероциклическими Li- производными. Так, реакция 2-тиениллития и 4-гексил-2-тиениллития с гексафторбензолом является первой стадией при получении олиготиофенов с центральным тетрафторфениленовым звеном (схема 101) [227].
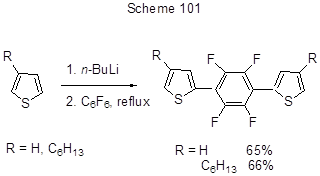
Бензодитиофены или -диселенофены, функционализированные концевыми полифторарильными группами, получены путём SNAr реакции их бис-литиевых производных и полифторарена (схема 102) [228].

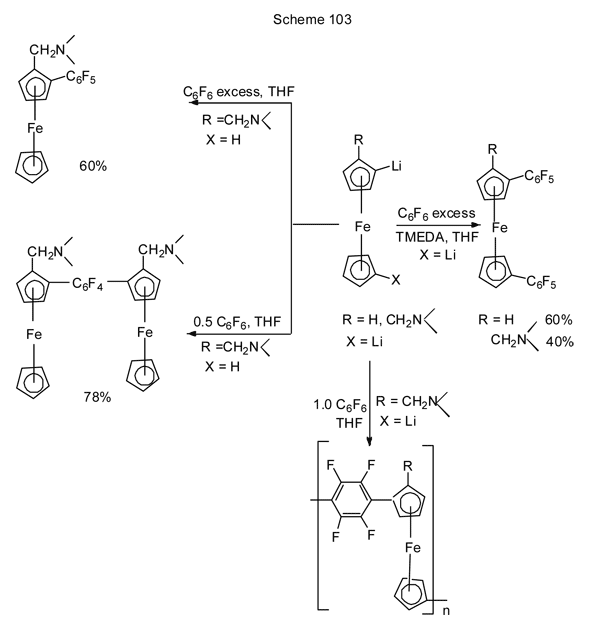
Описаны также реакции нуклеофильного замещения фтора в полифтораренах с Li-производными более сложных систем, таких, например, как ферроцен [229]. Гексафторбензол взаимодействует с бис- Li-производными ферроцена или диметиламинометилферроцена в присутствии TMEDA, давая бис-пентафторфенильные производные (схема 103) [229]. Аналогично монолитиевое производное (R = CH2N(CH3)2 , X = H) превращается с избытком гексафторбензола в моно-C6F5-производное, а в случае недостатка гексафторбензола получается 1,4-бис[2-(диметиламинометил)ферроцен-1-ил]тетрафторбензол [229].
Другой тип реакций с Li-органическими соединениями – активация связи СAr-F за счёт литирования орто-положения к атому фтора, что способствует отщеплению LiF и образованию соответствующего дегидробензола, способного к реакциям (4+2) циклоприсоединения. При наличии в орто-положении к реагирующему фтору атома брома и водорода литиирование идёт путём обмена на литий атома брома. В случае аналогичных хлорпроизводных обмен Н→Li более предпочтителен нежели Cl→Li. Для хлордифтораренов с галогенами в таких положениях, когда возможно образование двух разных дегидробензолов, например, для 1-хлор-2,4-дифторбензола, реализуется направление, в котором определяющим является индуктивный эффект атома хлора, который сильнее дестабилизирует положительный заряд на углероде в орто- положении, чем в пара- положении (схема 104) [230].

Такая региоселективность подтверждается получением соответствующих фторированных аддуктов фторсодержащих дегидробензолов, образующихся из ди-, три- и пентафторбром- или –хлораренов, в реакциях Дильса-Альдера с фураном или N-метилпирролом (схема 105) [230].
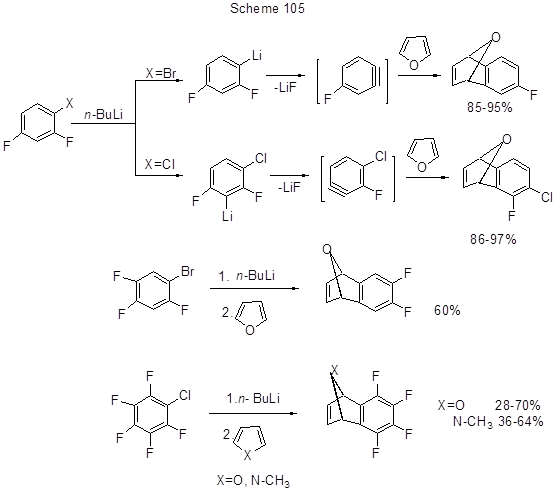
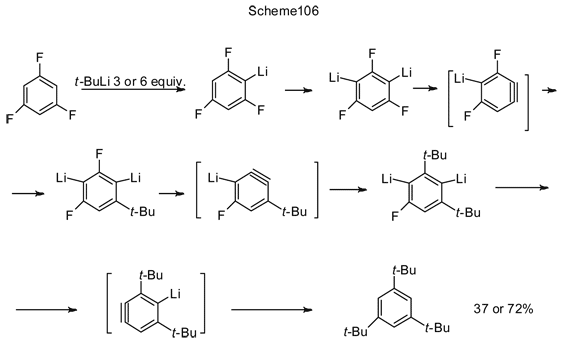
Ступенчатое литирование 1,3,5-трифторбензола вначале при использовании 3-х или 6-ти эквивалентных количеств t-BuLi продолжается каскадом альтернирующих отщеплений LiF и присоединением t-BuLi к генерирующимся дегидробензолам, приводя в результате к замещению на трет.-бутильную группу всех атомов фтора (схема 106) [231].
Приведённые выше реакции с участием дегидробензола проводились , как правило, в эфире, однако для реализации такого рода превращений могут быть использованы и некоординирующие, углеводородные растворители – пентан, гексан, гептан, дициклогексан, петролейный эфир [232]. Например, 2,6-дифториодбензол (1, схема 107) в петролейном эфире реагирует с n-BuLi и далее с циклопентадиеном, давая смесь двух соединений: 5-фтор-1,4-дигидро-1,4-метанонафталин 3 и 5-(2,6-дифторфенил)-1,4-дигидро-1,4-метанонафталин 4 [232].
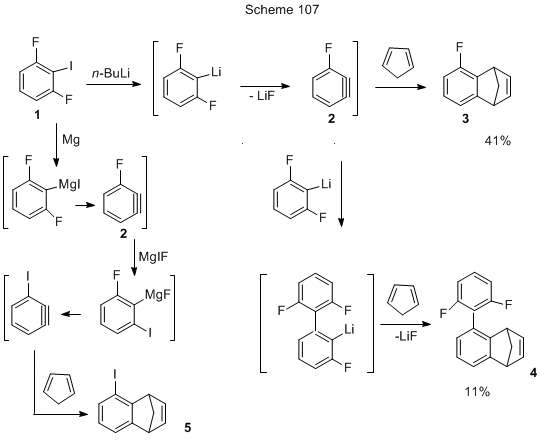
Дегидробензол 2 может быть генерирован из иодпроизводного 1 и через Mg-органическое соединение, но в этом случае в равном количестве с 3 получается иодпроизводное 5 при общем выходе смеси 41% [232].
1.3 Реакции с Mg-органическими производными
Магнийорганические соединения также активно использовались в качестве карбанионных источников при нуклеофильном замещении фтора в полифтораренах с целью введения желаемых С-группировок [233]. Так, в 2-(2,6-дифторфенил)-4,4-диметил-4,5-дигидро-1,3-оксазоле при действии CH3MgCl атом фтора в фенильном кольце меняется на CH3-группу с выходом 96% [233]. Аналогично вводилась метильная группа и в 1,3,4,5-тетрафторизофталонитрил или 1,3,4,5-тетрафторизофталевую кислоту, причём замещался атом фтора в пара-положении к CN- или COOH-группе (выходы 56% и 82% соответственно) [234]. Аналогичный результат получен при использовании в реакции с указанным нитрилом CH3MgBr. Реакция тетрафторизофталевой кислоты с 2-метилфенилмагнийхлоридом позволяет заместить атом фтора на 2-метилфенильную группу [234]. В плане получения и исследования жидко-кристаллических структур реализована реакция замещения пара-атома фтора пентафторфенильной группы в пентафторфенилтриметилсилилэтине на пара-алкоксифенильную группу при действии Mg-органического соединения из п-алкоксибромбензола [235]. Действие 3,4-Cl2C6H3MgCl или 3-CNC6H4MgCl на 2,3,4,5,6-пентафторбензофенон приводит к замещению на соответствующий арильный остаток атомов фтора в положениях 2 и 6 (выходы составляют 50% и 39% соответственно) [236]. В реакции октафторнафталина c C6F5MgBr получен с выходом 15% 2-пентафторфенилгептафторнафталин [237].
1.4 Действие Si-органических производных в присутствии катализаторов, генерирующих фторид-ион.
Для синтеза поли(фенилен)этиниленов с альтернирующими арил- и полифторарильными звеньями, которые могут представить большой интерес вследствие их оптоэлектронных свойств, предложен простой метод, альтернативный каталитической реакции Соногаширы, т.е. без использования соединений переходных металлов, ведущий к таким соединениям с высоким молекулярным весом, определённой структурой, чистотой и высоким выходом. Он состоит в нуклеофильном замещении атома фтора в полифторарене под действием Si-органических производных алкинаренов в присутствии катализаторов, генерирующих F- (CsF, TBAF, TMAF) (схема 108) [238]. Реакции идут при комнатной температуре. С пентафторпиридином превращение также протекает, но нужно использовать избыток фторида. В остальных случаях берётся его каталитическое количество. В модельном опыте (схема 108) главным является продукт замещения двух атомов фтора в пара- положении даже при соотношении гексафторбензола и триметилсилильного производного 1:1, что обусловлено большей скоростью замещения второго атома фтора по сравнению с первым.
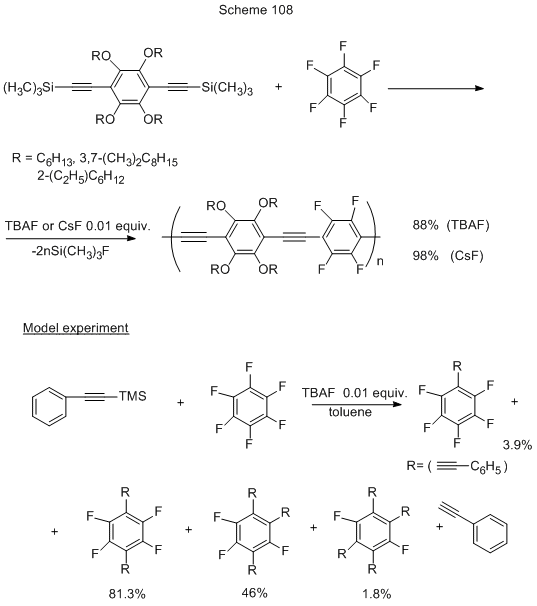
Другой пример использования Si-органических производных в присутствии F- см. на схеме 102 в разделе 1.2 [228].
2. Превращения по связи СAr-F, протекающие с её активацией.
Для успешного превращения связи СAr-F в СAr-C в ряде случаев требуется активация. Такая активация может достигаться различными путями. Об одном из таких путей (образование дегидробензола) уже говорилось выше в разделе 1.2. Активация может также достигаться применением микроволнового облучения или использованием различных катализаторов, в том числе органических. Так, региоспецифическое полифторарилирование гетероциклических аминалей кетенов успешно протекает при микроволновом облучении, как это показано на схеме 109 [239].
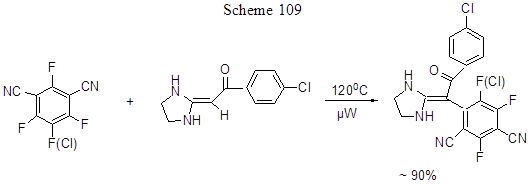
Нуклеофильное ароматическое замещение атома фтора в пара- положении к циан-группе в тетрафторизофталонитриле так же, как двух пара-атомов фтора в гексафторбензоле или пара-фтора в пентафторбензонитриле, идёт при действии 2,3-дигидро-1,3-диизопропил-4,5-диметил-имидазол-2-илидена в присутствии эфирата BF3. Выходы составляют более 80% [240]. Тетрафторфталонитрил при нагревании с КI в DMF с выходом 46% превращается в 2,2’,5,5’,6,6’-гексафтор-3,3’,4,4’-тетрациано-1,1’-бифенил [241]. Регио- и энантиоселективные SNAr реакции идут с активированными аренами и 1,3-дикарбонильными соединениями в условиях межфазного катализа с использованием четвертичной аммониевой соли из Cinchona Alkaloids [242, 243]. На схеме 110 представлена реакция циклоалкилирования нитрополифтораренов действием 2-карбэтоксициклопентанона или N-замещённого 3-карбэтокси-2-пирролидона в таких условиях. Замещение фтора идёт только в пара- положении к нитро-группе и тем легче, чем он более активирован. Механизм процесса включает отрыв основанием кислого протона в β-кетоэфире и генерацию амбидентного нуклеофила , который взаимодействует с хиральной четвертичной аммонийной солью, образуя хиральную ионную пару («хиральный карман»). Алкалоидная составляющая нуклеофила способствует его энантиоселективному нуклеофильному приближению к ароматическому субстрату.
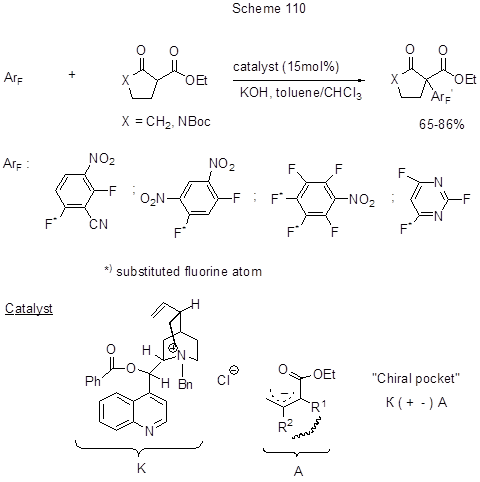
Превращение интересно как пример органокаталитической SNAr реакции. Однако, в последнее время в плане активации превращения СAr-F в CAr-C заметно большее значение и распространение получили методы, связанные с окислительным присоединением низковалентных металлов к С-F связи и дальнейшими трансформациями. Это широкий круг реакций кросс-сочетания, включаюший участие в качестве электрофилов фторароматических соединений, а нуклеофилов – металло- и элементоорганических соединений [реактивы Гриньяра (Кумада сочетание), борпроизводные (реакция Сузуки), Zn-реагенты (Негиши сочетание), Sn-реагенты (реакция Стилла)] в присутствии металлокомплексных катализаторов на основе Pd, Ni, Pt, Co. В обзоре [213] рассмотрены различные варианты таких реакций для фтораренов, в т.ч. и полифтораренов. Оказалось, что наиболее прочную из всех СAr-Hal связей - связь С-F - не так-то просто активировать и найти подходящие для этого комплексы переходных металлов. Так, связь СAr-F весьма селективно активируется в монофторбензолах действием Ni-комплексов [244], однако в случае гексафторбензола эти комплексы неактивны [245]. Тем не менее для полифторгетаренов была найдена система Ni(cod)2 + PEt3 (или РBu3), которая позволила провести алкенилирование пентафторпиридина и 2,3,5,6-тетрафторпиридина по реакции Стилла с Bu3SnCH=CH2 [246]. Реакция региоселективна и идёт только по положению 2 в обоих случаях. Предполагается, что прекоординация пиридинов по положению 2 с Ni-центром с образованием Ni-комплекса является определяющей ступенью в активации C-F связи (схема 111).
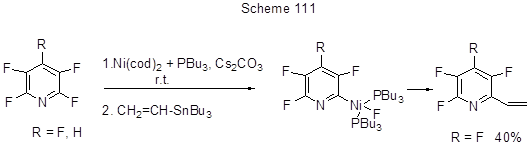
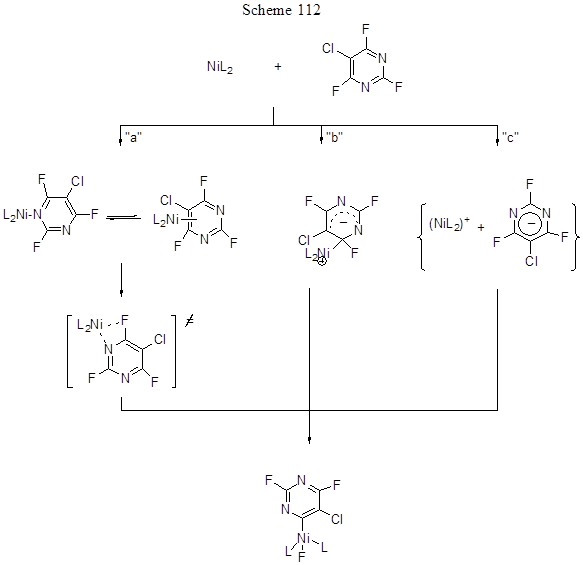
Побочные продукты – образование 4-Н-тетрафторпиридина из пентафторпиридина (причина –реакция последнего с триалкилфосфином) и 2,6-бис-винильного производного. Интересно, что связь СAr-H в 4-Н-тетрафторпиридине не участвует в реакции. Этот же катализатор, но с добавкой различных фосфинов PR3 , где R = Cy, i-Pr, Ph, был использован для арилирования 5-хлор-2,4,6-трифторпиримидина с ArB(OH)2 по реакции Сузуки, которая протекает по положениям 4,6 с выходом от 40 до 90%. Лучшее основание в этом случае - Cs2CO3 [244]. Существенно, что более слабая связь СAr-Cl при этом не затрагивается. Детальный анализ механизма превращения позволил авторам высказать предположение о трёх возможных путях образования Ni-комплекса (схема 112), хотя ни один из них не был подтверждён экспериментально.
Направление “а” включает прекоординацию пиримидина с никелем и далее – присоединение к окисляющемуся Ni-центру. Альтернативный путь “б” – это нуклеофильное замещение фтора, которое инициируется нуклеофильной атакой NiL2 по электрофильному положению ароматической системы как скорость определяющая стадия. Наконец, путь «с» – электронный перенос с образованием тесной ионной пары.
Использование другой каталитически активной системы никеля – NHC-стабилизированного Ni-комплекса (NHC- это N-гетероциклический карбен) [Ni(i-Pr2Im)4 (cod)] 1 (схема 113) [245], которая является источником [Ni(i-Pr2Im)2], было успешным и в ряду полифтораренов, таких как гексафторбензол, октафтортолуол и декафторбифенил и позволило реализовать реакцию арилирования по Сузуки. Реакция идёт с высокой региоселективностью с замещением атома фтора в пара-положении (схема 113).
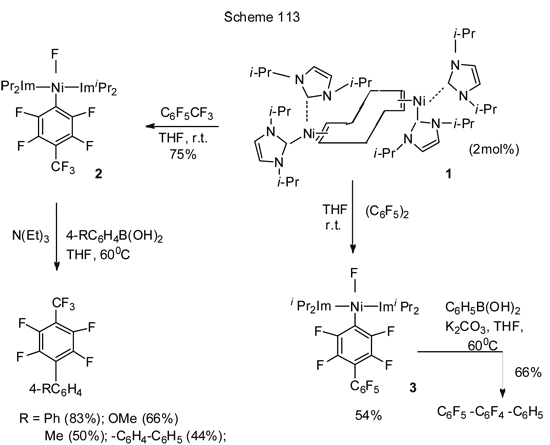
Каталитическая система 1 активна и в Кумада кросс-сочетании с реактивом Гриньяра, но выходы здесь обычно ниже. Так, 4-(4’-метилфенил)-гептафтортолуол из октафтортолуола и п-толилмагнийбромида получается по этой реакции только с выходом 26% [245]. В то же время реакция фенилирования методом Кумады изомерных дифторбензолов и 1,3,5-трифторбензола с PhMgBr и Ni-катализатором с триарилфосфиновым лигандом идёт настолько активно, что на фенильную группу замещаются все атомы фтора [247]. Реакция Кумады 2,4-дифторфенола с избытком n-С12Н25MgBr в присутствии NiCl2(diippbz) селективно протекает в орто-положение к гидроксильной группе, давая с выходом 75% соответствующий 2-алкил-4-фторфенол [248]. Такая региоселективность объясняется влиянием гидроксигруппы и тем, что в переходном состоянии стадии окислительного присоединения Ni льюисовско-кислый Mg облегчает разрыв связи C-F (рисунок 114), т.о. каталитичская Ni-система способствует орто-кросс-сочетанию дифторфенола с алкилгриньяровским реагентом.
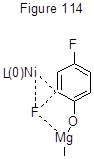
Селективное замещение орто-атома фтора в дифторзамещённых бензольных производных с электронодонорными заместителями (ОН, CH2OH, NH2, NHAc, NHBoc) с арилмагнийбромидами реализуется и при использовании палладиевого катализатора PdCl2(PCy3)2. Выходы 2-арильных производных в реакциях 2,4-дифторфенола достигают 80% и более [249]. Применение [PdCl2(dppf)] в реакции 1,2-дифторбензола с 4-CH3C6H4MgBr ведёт к замещению одного атома фтора на арильную группу, в то время как с Ni-катализатором NiCl2(dppp) получается смесь продуктов моно- и дифторзамещения примерно в равном количестве [5, p. 56]. Интересно отметить, что направление реакции Стилла пентафторпиридина с трибутилвинилоловом в присутствии палладиевого катализатора Pd[P(i-Pr)3]2 отличается от рассмотренного выше для Ni-катализатора (эта глава, схема 111). В случае палладивого катализатора получается Pd-комплекс в положении 4, который превращается в 4-винил-2,3,5,6-тетрафторпиридин [250].
Из катализаторов с другими переходными металлами, использованных в ряду фтораренов, можно отметить TaCl5 , который катализирует алкилирование гексафторбензола и пентафтортолуола действием 2-фенетилмагнийхлорида , приводящее к замещению пара-атома фтора, сопровождаемому перегруппировкой фенетильной группы (схема 115) [251].
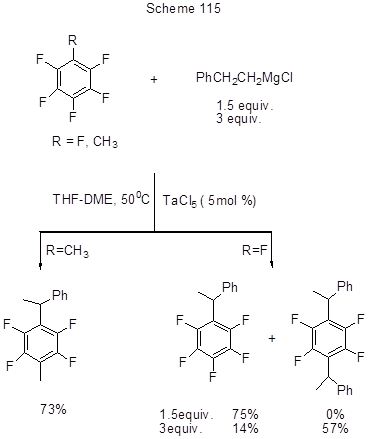
Интересно использование кобальтового катализатора в реакции Кумада полифтораренов, что позволяет устранить токсичность никилиевого и дороговизну палладиевого. Так, двухступенчатое орто,орто-арилирование пентафторбензофенона идёт при действии на него ArMgCl в присутствии Co(acac)2 и Cu(CN) . 2LiCl. Промоторами реакции являются Bu4NI и п-фторстирол (схема 116) [252]. В ходе реакции имеет место взаимодействие Mg-органического производного и соли меди с образованием в результате переметаллирования Cu-арена, который вступает в кросс-сочетание с пентафторбензофеноном, давая конечный продукт. В этой работе успешно продемонстрирована возможность вовлечения в реакции кросс-сочетания Cu-органических соединений, что менее исследовано, чем использование других металлорганических соединений в ряду арилфторидов для образования новой С-С связи.

Термическое разложение циркониевого комплекса Ср2Zr(C6F5)2 в присутствии фурана или дурола протекает с образованием аддуктов Дильса-Альдера, в то время как линейные перфторполифениленовые олигомеры пара-строения F-[-2,3,5,6-C6F4-]n-F, (n=2-13) получаются при термическом разложении этого комплекса в присутствии гексафторбензола или декафторбифенила [253]. Наблюдаемые закономерности согласуются с радикальным цепным процессом в случае полифтораренов, который инициируется примесями в этих аренах и реализуется в первой, быстрой стадии разложения комплекса, а также и механизмом с промежуточным образованием тетрафтордегидробензола, который ответственнен за образование аддуктов Дильса-Альдера в случае фурана и дурола.
Активация связи СAr-F в полифтораренах с образованием полифторарилцинкорганических соединений идёт при действии Zn в присутствии SnCl2 в DMF [254, 184]. Образующиеся Zn-органические производные нагреванием с CuCl2 превращаются в полифторбиарилы (схема 117) [254], а их реакция с аллилхлоридом в системе CuCl/DMF приводит к образованию аллильных производных [184]. Селективное орто-метилирование полифторированных арилиминов, содержащих в ароматическом кольце от двух до пяти атомов фтора, по связи СAr-F происходит при действии на них диметилцинка в присутствии платинового катализатора Pt2Me4(SMe2)2 (схема 118) [255]. В случае 2,6-дифтор-4-бромпроизводного такая региоселективность схраняется, несмотря на наличие в молекуле более слабой связи СAr-Br.
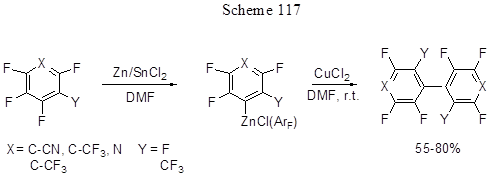
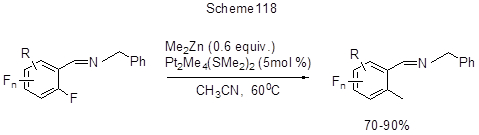
Активация орто-атома фтора идёт за счёт образования циклического Pt-комплекса с участием атома азота и орто-CAr-F связи. В платиновом комплексе под действием Me2Zn происходит переметаллирование, а последующее восстановительное элиминирование приводит к образованию конечного продукта и регенерации Pt(II)-катализатора.
V. Превращение других заместителей в полифторарене
Введение алкильных, арильных (гетарильных) и непредельных группировок в полифторарен возможно не только с участием связи СAr-H или CAr-Hal, где Hal=Br,Cl,F,I, но и путём трансформации других заместителей, имеющихся в молекуле. Так, трансформация С=О связи в полифторарилкетонах приводит к соответствующим гидроксиалкильным производным (схема 119) [256-258].

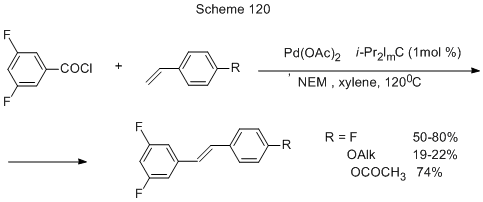
Сочетанием 3,5-дифторбензоилхлорида с замещёнными стиролами по декарбонилативной реакции Мизороки-Хека получаются полифторированные стильбены (схема 120) [259].
Подобного рода декарбоксилативная реакция гетарилирования 2,6-дифторбензойной кислоты действием N-замещённого индола позволяет вводить вместо карбоксильной группы индольный фрагмент (схема 121) [260]. В этом превращении Pd-катализатор активирует 3-е положение индола, а соль серебра способствует декарбоксилированию кислоты.
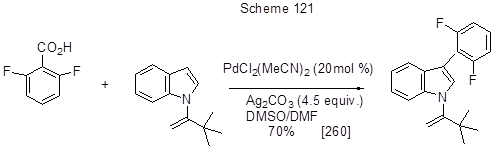
Декарбоксилативное кросс-сочетание К-солей ди-, три-, тетра- и пентафторбензойных кислот с различными иод- и бромпроизводными бензола, пиридина и тиофена реализуется только при использовании медного катализа (CuI или CuI + Phen) (схема 122) [261].
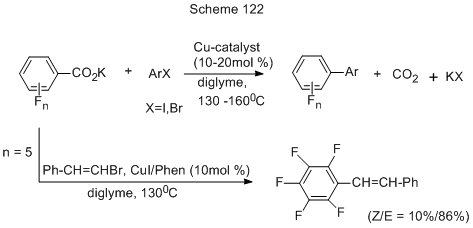
В этой методике не используются обычные дорогие и нестабильные металлорганические катализаторы, и в результате вместо токсичных галогенидов металлов образуется СО2. Метод позволяет получать полифторированные биарилы и дополняет описанные в предыдущих разделах. Иодиды реагируют с высокими выходами при использовании CuI с лигандом или без лиганда, а для бромидов присутствие лиганда обязательно. Использованные галогенопроизводные содержали как электронодонорные, так и электроноакцепторные заместители в орто-, мета- и пара-положениях. На примере реакций бромпроизводных высказано предположение, что механизм реакции (схема 123) включает первоначальное взаимодействие катализатора [иодмедного комплекса (1)] и К-соли кислоты с образованием Сu-соли кислоты (2), которая через 4х-членное циклическое переходное состояние (TS 2-3) декарбоксилируется и превращается в новый медный комплекс (3), содержащий полифторароматический фрагмент. Этот медный комплекс взаимодействует с арилбромидом путём окислительного присоединения через переходное состояние (TS 3-4), давая новый медный комплекс с полифторарильным и арильным фрагментом (4), который в результате восстановительного элиминирования через переходное состояние (TS 4-5) превращается в конечный биарил.

При использовании в этой реакции бромстиролов могут быть получены полифторированные стильбены [261]. 3,3',5,5' –Тетрафторстильбен образуется также в реакции (3,5-дифторбензил)трифенилфосфонийбромида с 3,5-дифторбензальдегидом (схема 124) [262].
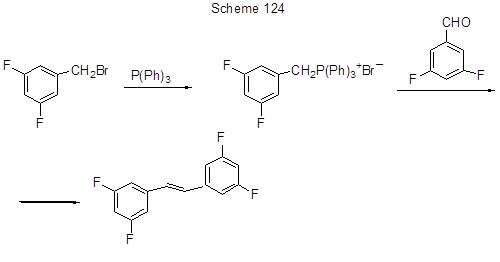
Образование винильного фрагмента в 5-ом положении 3,4-дифтор-5-гидроксиалкилпроизводного толуола идёт за счёт дегидратации (схема 125) [85], а реакция бензилирования аренов – удобный путь к диарилметанам, исходя из бензиловых спиртов (схема 126) [263].
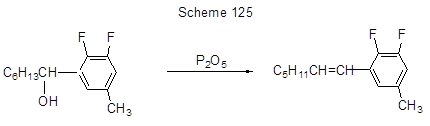
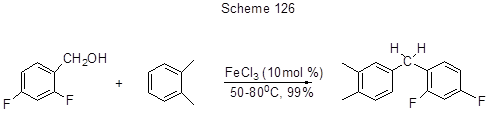
Двуcтупенчатый синтез 1-дифторфенил-2-пропанонов из дифторанилинов и изопропилацетата по реакции Мейервейна предложен в работе [264] (схема 127).
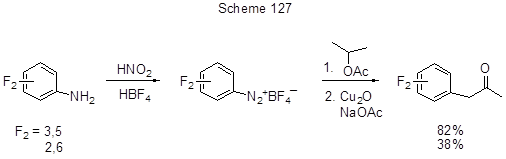
VI. З А К Л Ю Ч Е Н И Е
Исследования по алкилированию, арилированию (гетарилированию), алкенилированию и алкинилированию
полифтораренов показывают, что как и в нефторированном ряду, для этих соединений наряду с классическими
методами проведения таких реакций всё большее распространение получают методы с использованием металлокомплексного
катализа, которые при подборе соответствующих металлов и лигандов позволяют с успехом вводить нужные
группировки в полифторированный арен как путём замещения атомов водорода, так и любого галогена,
включая атом фтора. Эти превращения часто являются одной из стадий в многостадийном синтезе и во
многих случаях используются для получения соединений с практически полезными свойствами. Развитие
таких методов в ряду полифтораренов безусловно будет способствовать расширению числа соединений,
содержащих полифторароматическую группировку, исследованию их химических свойств и поиску путей их
практического применения.
Список сокращений
Список литературы
[2] Mоррисон Р., Бойд Р. Органическая химия., Мир, Москва, (1974).
[3] Бартон Д., Оллис В.Д. Общая органическая химия, Т.1., Химия, Москва, (1981).
[4] Hassan J., Sevignon M., Gozzi C., Schulz E., Lemair M., Chem. Revs., (2002), 102, 1359-1470.
[5] Ed. Ackermann L. Modern Arylation Methods., John Wiley, Weinheim, (2009).
[6] Смит В.А., Дильман А.Д. Основы современного органического синтеза., БИНОМ, Лаборатория знаний, Москва, (2009).
[7] Eds. Meijere A., Diederich F. Metal-Catalyzed Cross-Coupling Reactions., John Wiley, Weinheim, (2004).
[8] Schlosser M. Organometallics in Synthesis, 2nd Ed., John Wiley, Chichester, (2002).
[9] Brooke G.M., J. Fluor. Chem., (1997), 86, 1-76.
[10] Filler R., Fiebig Jr. A.E., Mandal B.K., J. Fluor. Chem., (2000), 102, 185-188.
[11] Koda T., Yasntake M., Shinmyozu T., Org. Lett., (2001), 3, 1419.
[12] Tabart M., Picaut G., Desconclois J-F., Dutka-Maben S., Huet Y., Berthaud N., Bioorg. Med. Chem. Lett., (2001), 11, 919-922.
[13] Lundbeck H., Pat. WO 106534 (2009) (C.A., 2009, 151, 337044).
[14] Nakamura S., Sugimoto H., Ohwada T., J. Am. Chem. Soc., (2007), 129, 1724- 1732.
[15] Pat. US 211741 (2006) (C.A., 2006, 145, 377029).
[16] Pat. US 270682 (2006) (C.A., 2007, 146, 27833).
[17] Карпов В.М., Меженкова Т.В., Платонов В.Е., Синяков В.Р., Щёголева Л.Н., Ж. орган. хим., (2002), 38, 1210-1217.
[18] Karpov V.M., Meshenkova T.V., Platonov V.E., Sinyakov V.R., J. Fluor.Chem., (2002), 117, 73-81. Karpov V.M., Meshenkova T.V., Platonov V.E., Sinyakov V.R.,Shchegoleva L.N., Russ. J. Org. Chem.,(2002), 38, 1158-1165 (English translation).
[19] Синяков В.Р., Меженкова Т.В., Карпов В.М., Платонов В.Е., Рыбалова Т.В., Гатилов Ю.В., Ж. орган. хим., (2003), 39, 886-891. Sinyakov V.R., Meshenkova T.V., Karpov V.M., Platonov V.E., Rybalova T.V., Gatilov Y.V., Russ. J. Org. Chem.,(2003),39, 837-842 (English translation).
[20] Zonov Ya.V., Karpov V.M., Platonov V.E., J. Fluor.Chem., (2007), 128, 1058- 1064.
[21] Синяков В.Р., Меженкова Т.В., Карпов В.М., Платонов В.Е., Ж. орган. хим., (2007), 43, 1678-1686. Sinyakov V.R., Meshenkova T.V., Karpov V.M., Platonov V.E., Russ. J. Org. Chem.,(2007),43, 1677-1685 (English translation).
[22] Karpov V.M., Mezhenkova T.V., Platonov V.E., Sinyakov V.R., J. Fluor. Chem., (2001), 107, 53-57.
[23] Синяков В.Р., Меженкова Т.В., Карпов В.М., Платонов В.Е., Рыбалова Т.В., Гатилов Ю.В., Ж. орган. хим., (2006), 42, 85-93. Sinyakov V.R., Meshenkova T.V., Karpov V.M., Platonov V.E., Rybalova T.V., Gatilov Y.V., Russ. J. Org. Chem.,(2006),42, 77-86 (English translation).
[24] a) Bonnefous C., Payne J.E., Roppe J., Zhuang H., Chen X., Severance D., Noble S.A., Smith N.D., Symons K.T., Nguyen P.M., Shiau A.K., Hassig C.A., Sablad M., Rosenkrants N., Zhang Y., Tadimeti S., Wang L., Rix P., Walsh J.P., Yazdani N., J. Med. Chem., (2009), 52, 3047-3062; b) Pat. WO 29592 (2009) (C.A., 2009, 150, 283051); c) Pat. WO 29617 (2009) (C.A., 2009, 150, 282846); d) Pat. WO 29625 (2009) (C.A., 2009, 150, 283049).
[25] Safina L.Yu., Selivanova G.A., Shteingarts V.D., Koltunov K.Yu., Tetrahedron Letters., (2009), 50, 5245-5247.
[26] Inglis S.R., Stojkoski C., Branson K.M., Cawthray J.F., Fritz D., Wiadrowski E., Pyke S.M., Booker G.W., J. Med. Chem., (2004), 47, 5405-5417.
[27] Breault G., Eyemann C.J., Geng B., Momingstar M., Reck F., Pat. WO 134378 (2006) [C.A. (2007), 146, 81779].
[28] Kato T., Saeki K., Kawazoe Y., Hakura A., Mutation Res., (1999), 439, 149- 158. [29] Laev S.S., Gurskaya L.Yu., Selivanova G.A., Beregovaya I.V., Shchegoleva L.N., Vasil’eva N.V., Shakirov M.M., Shteingarts V.D., Eur. J. Org. Chem., (2007), 306-316.
[30] Shuttleworth S.J., Lizarzabaru M.E., Chai A., Coward P., Bioorg. Med. Chem. Letters., (2004), 14, 3037-3042.
[31]Мустафин А.Г.,Гимадиева А.Р.,Халилов И.Н.,Спирихин Л.В.,Фатыхов А.А.,Нурушев Р.А.,Абрахманов И.Б.,Толстиков Г.А.,Изв.АН.Сер.хим., (1998), 188-190. Mustafin A.G., Gimadieva A.R., Khalilov I.N., Spirikhin L.N., Fatykhov A.A., Nurushev R.A., Abdrakhmanov I.B., Tolstikov G.A., Bull. Russ. Acad. Sci. Div. Chem.Sci., (1998), 47, 188-190 (English translation).
[32]Гатауллин Р.Р.,Миннигулов Ф.Ф.,Кудашев А.Р.,Нурушев Р.А.,Абрахманов И.Б.,Ж.прикл.хим., (2002), 75, 95-97. Gataullin R.R., F. F. Minnigulov F.F., Kudashev A.R., Nurushev R.A., I. B. Abdrakhmanov I.B., Russ. J. Applied Chem., (2002), 75, 95-97 (English translation).
[33]Гатауллин Р.Р.,Кажанова Т.В.,Давыдова В.А.,Исмагилова А.Ф.,Зарудный Ф.С.,Фатыхов А.А.,Спирихин Л.В.,Абрахманов И.Б.,Хим.-фарм.ж. (1999), 33, 29-32. Gataullin R.R., Kazhanova T.V., DavydovaV.A., IsmagilovaA.F., Zarudii F.S., Fatykhov A.A., SpirikhinL.V., I.B. AbdrakhmanovI.B., Russ. J. Chem. Pharm., (1999), 33, , 255-258.
[34] Song Yu., Zhu A., Lv J., Gong G., Xie J., Zhou J., Ye Y., Zhong X., Spectrochim. Acta Part A: Molecular and Biomol. Spectroscopy, (2009), 73, 96-100.
[35] Chackalamannil S., Doller D., Clasty M., Xia Y., Fagen K., Lin Y., Hsung-An Tsai, McPhail A.T., Tetrahedron Letters, (2000), 41, 4043-4047.
[36] Yoshimura H., Kikuchi K., Hibi Sh., Tagami K., Satoh T., Yamauchi T., Ishibahi A., Tai K., Hida T., Tokuhara N., Nagai M., J. Med. Chem., (2000), 43, 2929-2937.
[37] Pat. US 6110959 (2000) (C.A., 1997, 127, 278135); Pat. US 6355669 (2002) (C.A., 1999, 130, 306588); Pat. US 6258811 (2001) (C.A., 1998, 128, 270532).
[38] Pat. US 7045545 (2006) (C.A., 2000, 133, 135218).
[39] Kim B.H., Jeon I., Lee D.B., Park H.J., Jun Y.M., Baik W., J. Fluor. Chem., (2001), 109, 145-149. 106
[40] Yekta S., Krasnova L.B., Mariampillai B., Picard C.J., Chen G., Pandiaraju S., Yudin A.K., J. Fluor. Chem., (2004), 125, 517-525; Pat. WO 51899 (2005) (C.A., 2005, 143, 27355).
[41] Pat. WO 60160 (2009) (C.A., 2009, 150, 313452).
[42] Pat. US 78249 (2003) (C.A., 2001, 134, 86149).
[43] Lafrance M., Rowley Ch.N., Woo T.K., Fagnou K., J. Am. Chem. Soc., (2006), 128, 8754-8756.
[44] Lafrance M., Shore D., Fagnou K., Org. Letters, (2006), 8, 5097-5100.
[45] Youbert N., Urban M., Pohl R., Hocek M., Synthesis, (2008), 1918-1932.
[46] Litvinas N.D., Brodsky B.H., DuBois J., Angew. Chem., Int. Ed.Engl., (2009), 48, 4513-4516.
[47] Zhao D., Wang W., Lian S., Yang F., Lan J., You J., Chemistry – A Europ. J., (2009), 15, 1337-1340.
[48] Itahara T., J. Org. Chem., (1985), 50, 5546-5550.
[49] Ahn J.H., Cho S.Y., Ha J-D., Chu S.Y., Jung S.H., Jung Y.S., Baek J.J., Choi I.K., Shin E.Y., Kang S.L., Kim S.S., Cheon H.G., Yang S.D., Choi J.K., Bioorg. Med. Chem. Letters, (2002), 12, 1941-1946.
[50] Rauf W., Thompson A.L., Brown J.M., Chem. Commun., (2009), 3874-3876.
[51] Laha J.K., Petron Ph., Cuny G.D., J. Org. Chem., (2009), 74, 3152-3155.
[52] Chernyak N., Gevorgyan V., J. Am. Chem. Soc., (2008), 130, 5636-5637; Advanced Synth. and Catalysis, (2009), 351, 1101-1114.
[53] Wei Y., Kan J., Wang M., Li W., Hong M., Org. Letters, (2009), 11, 3346- 3349.
[54] Demchuk O.M., Pietrusiewicz K.M., Synlett, (2009), 18, 1149-1153.
[55] Pat. US 42896 (2009) (C.A., 2009, 150, 237419).
[56] Saito A., Oda S., Fukaya H., Hanzawa Yu., J.Org. Chem., (2009), 74, 1517- 1524.
[57] Nakao Y., Kashihara N., Kanyiva K.S., Hiyama T., J. Am. Chem. Soc., (2008), 130, 16170-16171.
[58] Pat. US 6211375 (2001) (C.A., 1997, 127, 331407).
[59] Pat. US 6136823 (2000) (C.A., 1998, 129, 41087).
[60] Pat. US 6235764 (2001) (C.A., 2000, 132, 22961).
[61] Pat. US 6348624 (2002) (C.A., 1999, 131, 73434); Pat. US 6329391 (2001) (C.A., 2002, 136, 20031).
[62] Pat. EP 1223169 (2002) (C.A., 1996, 125, 142540).
[63] Pat. US 114666 (2003) (C.A., 2003, 138, 55976).
[64] Chodakowski J., Klis’ T., Servatowski J., Tetrahedron Letters, (2005), 46, 1963-1965.
[65] Anquetin G., Greiner J., Vierling P., Tetrahedron, (2005), 61, 8394-8404.
[66] Omura K., Uchida T., Irie R., Katsuki T., Chem. Commun., (2004), 2060- 2061.
[67] Leroux F., Hutsehenreuter T.U., Charriere C., Scopelliti R., Hartman R.W., Helv. Chim. Acta, (2003), 86, 2671-2686.
[68] Inagaki H., Tsuruoka H., Hornsby M., Lesley S.A., Spraggon G., Ellman J.A., J. Med. Chem., (2007), 50, 2693-2699.
[69] Bashore C.G., Vetelino M.G., Wirtz M.C., Brooks P.R., Frost H.N., McDermort R.E., Whritenour D.C., Ragan J.A., Rutherford J.L., Makowski T.W., Brenek S. J., Coe J.W., Org. Letters, (2006), 8, 5947-5950.
[70] Becht J.M., Ngouela S., Wagner A., Mioskowski Ch., Terahedron, (2004), 60, 6853-6858.
[71] Leroux F.R., Bonnafoux L., Heiss C., Colobert F., Lanfranchi D.A., Advanced Synth. and Catalysis, (2007), 349, 2705-2713.
[72] Milne J.E., Buchwald S.L., J. Am. Chem. Soc., (2004), 126, 13028-13032.
[73] Heis C., Leroux F., Schlosser M., Eur. J. Org. Chem., (2005), 5242-5247.
[74] Pat. US 6388146 (2002) (C.A., 1999, 131, 163453).
[75] Scott J.P., Brower S.E., Davis A.J., Brands K.M.J., Synlett, (2004), 1646-1648.
[76] Davies A.J., Scott J.P., Bishop B.C., Brands K.M., Brewer S.E., DaSilva J.O., Dormer P.G., Dolling U-H., Gibb A.D., Hammond D.C., Lieberman D.D., Palucki M., Payack J.T., J. Org. Chem., (2007), 72, 4864-4871.
[77] Pat. US 42851 (2009) (C.A., 2007, 147, 234875).
[78] Nishida J., Fujiwara Yu., Yamashita Y., Org. Letters, (2009), 11, 1813-1816.
[79] Pat. US 131482 (2009) (C.A., 2009, 150, 563826).
[80] Chen Sh., Huby N.J.S., Kong N., Moliterni J.A., Jonh A., Morales O.J., Pat. US 170920 (2009) (C.A., 2009, 151, 123990).
[81] Keum H.-W., Roh S.D., Do Y.-S., Lee J.-H., Kim Y.-B., Mol. Crystals and Liquid Crystals, (2005), 439, 189-199.
[82] Bremer M., Lietrau L., New J. Chem., (2005), 29, 72-74.
[83] Pat. US 247585 (2007) (C.A., 2007, 147, 494408).
[84] a) Glendenning M.E., Goodby J.W., Hird M., Toyne K.J., J. Chem. Soc., P2, (2000), 27-34; b) J. Chem. Soc., P2 (1999), 481-491.
[85] Cammidge A.N., Crepy K.V.L., J. Org. Chem., (2003), 68, 6832-6835. [86] Popov I., Do H-Q., Daugulis O., J. Org. Chem., (2009), 74, 8309-8313.
[87] Coe P.L., Rees A.J., J. Fluor. Chem., (2000), 101, 45-60.
[88] Do H.-Qu., Daugulis O., J. Am. Chem. Soc., (2008), 130, 1128-1129.
[89] Do H.-Qu., Khaw R.M.K., Daugulis O., J. Am. Chem. Soc., (2008), 130, 15185-15192.
[90] Pat. US 76266 (2009) (C.A., 2009, 150, 329777).
[91] Do H.-Qu., Daugulis O., Chem. Commun., (2009), 6433-6435.
[92] Matsuyama N., Kitahara M., Hirano K., Satoh T., Miura M., Org. Letters, (2010), 12, 2358-2361.
[93] Susuki A., J. Organomet. Chem., (1999), 576, 147-168.
[94] Molander G.A., Biolatto B., J. Org. Chem., (2003), 68, 4302-4314.
[95] Kotsikorou E., Song Y., Chan J.M.W., Faelens S., Tovian Z., Broderick E., Bakalara N., Docampo R., Oldfield E., J. Med. Chem., (2005), 48, 6128-6139.
[96] Song Y., Oldfield E., Lin F.-Y., Yin F, Mikkamala D., Dushyant C., Cao R., Hensler M., Nizet V., Poveda C-A.R., Pacanowska D.G., Wang H., Morita C.T., J. Med. Chem., (2009), 52, 976-988.
[97] Kumar M., Kaur K., Sinha S., Gupta S., Palle V., Chugh A., Pat US 12116 (2009) (C.A., 2007, 146, 162865).
[98] Feuerstein M., Berthiol F., Doucet H., Santelli M., Synlett, (2002), 1807-1810.
[99] Kulmaelae T., Kuuloja N., Franzen R.X., Rissanen R.K., Europ. J. Org. Chem., (2008), 4019-4024.
[100] Guo M., Zhang Q., Tetrahedron Letters, (2009), 50, 1965-1968.
[101] Duplantier A.J., Kraus K.G., Lu J., Noha M., Rogers B.N., Zhang L., Efremov I., Candler J., O’ Sillivan T.J., Ganong A.H., Hanks A.N., Lazzaro Jr. J.T., McCarthy S.A., Siuciak J.A., Doran A.C., Haas J.A., Spracklin D.K., Bioorg. Med. Chem. Letters, (2009), 19, 2524-2529.
[102] Pat. EP 1842854 (2007) (C.A., 2007, 147, 427511); Pat. WO 148829 (2008) (C.A., 2009, 150, 44027); Pat. US 200920 (2009) (C.A., 2007, 146, 430964).
[103] Pat. WO 11447 (2009) (C.A., 2009, 150, 121792).
[104] Pat. US 163508 (2009) (C.A., 2009, 150, 447978). 109
[105] Pat. WO 112854 (2009) (C.A., 2009, 151, 392217).
[106] Pat. WO 17664 (2009) (C.A., 2009, 150, 191528); Pat. WO 10150 (2010) (C.A., 2010, 152, 215298).
[107] Pat. US 203705 (2009) (C.A., 2009, 151, 245649).
[108] Pat. WO 155388 (2009) (C.A., 2010, 152, 97501); Pat. WO 155389 (2009) (C.A., 2010, 152, 97472).
[109] Pat. WO 102460 (2009) (C.A., 2009, 151, 289162).
[110] Pat. WO 153285 (2009) (C.A., 2010, 152, 97447).
[111] Ce G., Guo H., He J., Wang F., Zou D., J. Organomet. Chem., (2009), 694, 3050-3057.
[112] Lumeras W., Caturla F., Vidal L., Esteve C., Vidal B., Balaque C., Orellana A., Godessard N., Dominguez M., Roca R., Huerta J.M., J. Med. Chem., (2009), 52, 5531-5545.
[113] Imbriglio J.E., Chang S., Liang R., Raghavan S., Schmidt D., Smenton A., Tria S., Schrader T.O., Jung J.K., Esser C., Taggart A.K.P., Cheng K., Carballo-Jane E., Waters M.G., Tata J.P., Colletti S.L., Bioorg. Med. Chem. Letters, (2009), 19, 2121-2124.
[114] Matin A., Gavandl N., Kim M.S., Yang N.X., Salam N.K., Hanzahan J-R., Roubin R.H., Hibbs D.E., J. Med. Chem., (2009), 52, 6835-6850.
[115] Pat. WO 26657 (2009) (C.A., 2009, 150, 306367).
[116] Hagdar S.N., Comery T.A., Dunlop J., Ghiron Ch., Bettinetti L., Bochmann H., LaPosa S., Micco I., Pollastrini M., Quinn J., Roncarati R., Scali C., Valacchi M., Varrone M., Zanaletti R., Bioorg. Med. Chem., (2009), 17, 5247-5258.
[117] Lee S., Yi K.Y., Youn S.J., Lee B.A., Yoo S., Bioorg. Med. Chem. Letters, (2009), 19, 1329-1331.
[118] Pat. WO 52341 (2009) (C.A., 2009, 150, 472421).
[119] Blake T.D., Hamper B.C., Huang W., James R., Moon J.B., Neel B.E., Olson K.L., Pelc M.J., Schneitzer B.A., Thorarensen A., Trujillo J.I., Turner S.R., Pat. US 146569 (2008) (C.A., 2008, 149, 79492).
[120] Pat. US 170907 (2009) (C.A., 2007, 146, 62449).
[121] Burns M.J., Fairlamb I.J.S., Kapdi A.R., Sehnal P., Taylor R.J.K., Org. Letters, (2007), 9 , 5397-5400; Fairlamb I.J.S., Sehnal P., Taylor R.J.K., Synthesis, (2009), 508-510.
[122] Pat. EP 2123623 (2009) (C.A., 2008, 149, 235600). 110
[123] a) Pat WO 120789 (2009) (C.A., 2009, 151, 403339); b) Pat. EP 2116522 (2009) (C.A., 2008, 149, 343403).
[124] Li G., Stamford A.W., Huang Y., Cheng K.Ch., Cook J., Farley C., Gao J., Ghibaudi L., Greenlee W.J., Guzzi M., van Heek M., Hwa J.J., Kelly J., Mullins D., Parker E.M., Wainhaus S., Zhang X., Bioorg. Med. Chem. Letters, (2008), 18, 1146- 1150.
[125] Bouillot A.M., Dodic N., Gellibert F.J., Mirguet O., Pat. WO 15652 (2010) (C.A., 2010, 152, 238948).
[126] Demir A.S., Findik H., Saygili N., Subari N.T., Tetrahedron, (2010), 66, 1308-1312.
[127] Dong Ch.Ch., Styring P., Goodby J.W., Chan L.K.M., J. Mater. Chem., (1999), 9, 1669-1678.
[128] Yelamaggad C.V., Mathews M., Hizemath U.S., Rao D.S., Prasad S.K., Tetrahedron Letters, (2005), 46, 2623-2626.
[129] Deeg O., Bauerle P., Org. and Biomol. Chem., (2003), 1, 1609-1624.
[130] Pat. WO 52722 (2006) (C.A., 2006, 144, 488936).
[131] Tomson S.A., Banker P., Bickett D.M., Boucheron J.A., Carter H.L., Clancy D.C., Cooper J.P., Dickerson S.H., Garrido D.M., Nolte R.T., Peat A.J., Sheckler L.R., Sparks S.M., Tavares F.X., Wang L., Wang T.Y., Weiel J.E., Bioorg. Med. Chem. Letters, (2009), 19, 1177-1182.
[132] Wood W.J.L., Patterson A.W., Tsuruoka H., Jain R.K., Ellman J.A., J. Am. Chem. Soc., (2005), 127, 15521-15527.
[133] Pat. WO 77385 (2009) (C.A., 2009, 151, 77775).
[134] Liu P., Zhou L., Li X., He R., J. Organomet. Chem., (2009), 694, 2290-2294.
[135] Bey E., Marchais-Oberwinkler S., Negri M., Kruchten P., Oster A., Kllin T., Spadaro A., Werth R., Frotscher M., Birk B., Hartmann R.W., J. Med. Chem., (2009), 52, 6724-6743.
[136] Pat. WO 45383 (2009) (C.A., 2009, 150, 396692).
[137] Pat. US 258866 (2009) (C.A., 2008, 148, 17746).
[138] Jaroch S., Hölscher P., Rehwinkel H., Sülzle D., Burton G., Hillmann M., McDonald F.M., Bioorg. Med. Chem. Letters, (2002), 12, 2561-2564; Jaroch S., Rehwinkel H., Hölscher P., Sülzle D., Burton G., Hillmann M., McDonald F.M., Miklautz H, Bioorg. Med. Chem. Letters, (2004), 14, 743-746.
[139] Zhou L., Du X., He R., Ci Z., Bao M., Tetrahedron Letters, (2009), 50, 406- 408.
[140] Stencel L.M., Kormos C.M., Avery K.S., Leedbeater N.E., Org. Biomol. Chem., (2009), 7, 2452-2457.
[141] Pat. US 239748 (2009) (C.A., 2007, 146, 121670).
[142] Pat. EP 2011788 (2009) (C.A., 2007, 147, 502372).
[143] Barberousse V., Bondoux M., Tomas D., Peyrou V., Pat. US 293768 (2008) (C.A., 2006, 145, 377529); Bondoux M., Mignon L., Ou K., Renaut P., Tomas D., Barberousse V., Tetrahedron Letters, (2009), 50, 3872-3876.
[144] Eidenschink R., Kretzchmann H., Pat. US 131689 (2009) (C.A., 2007, 147, 200299).
[145] Pat. WO 91884 (2009) (C.A., 2009, 151, 208832).
[146] Pat. US 29935 (2010) (C.A., 2008, 148, 403569).
[147] Lugo B., Allbutt B., Beaumont D., Butt U., James A.R., Synlett, (2009), 675- 680.
[148] Taugerbeck A., Montenegro E., Manabe A., Plach H., Pat US 247620 (2009) (C.A., 2007, 147, 177293).
[149] Hird M., Goodby J.W., Gough N., Toyne K.J., J. Mater. Chem., (2001), 11, 2732-2742.
[150] Raghavan S., Schmidt D.R., Darby R., Steven S.L., Abi- Smenton A.L., Pat. US 62269 (2009) (C.A., 2007, 147, 277179).
[151] Korenaga T., Kosaki T., Fukumura R., Ema T., Sakai T., Org. Letters, (2005), 7, 4915-4917.
[152] a) Frohn H-J., Adonin N. Yu., Bardin V.V., Starichenko V.F., J. Fluor. Chem., (2003), 122, 195-199; b) Tetrahedron Letters, (2002), 43, 8111-8114; c) J. Fluor. Chem., (2002), 117, 115-120.
[153] Pat. WO 109743 (2009) (C.A., 2009, 151, 337021).
[154] Levi M.D., Gofer Y., Cherkinsky M., Birsa M.L., Aurbach D., Berlin A., Phys. Chem. Chem. Phys., (2003), 5, 2886-2893.
[155] Mosrin M., Knochel P., Org. Letters, (2009), 11, 1837-1840.
[156] Pat. EP 987238 (2000) (C.A., 1998, 129, 223634).
[157] Wunderlich S.H., Knochel P., Angew. Chem., Int. Ed.Engl., (2009), 48, 1501- 1504.
[158] Bromidge S.M., Dabts S., Davies D.T., Davies S., Duckworth D.M., Forbes I.T., Gaster L.M., Ham P., Jones G.E., King F.D., Mulholand K.R., Saunders D.V., Wyman P.A., Blaney F.E., Clarke S.E., Blackburn T.P., Holland V., Kennett G.A., Lightower S., Middlemiss D.N., Trail B., Rilly G.J., Wood M.D., J. Med. Chem., (2000), 43, 1123-1134. [158a] Pat. WO 14268 (2009), (C.A., (2009), 150, 213983).
[159] a) Hatakeyama T., Hashimoto S., Ishizuka K., Nakamura M., J. Am. Chem. Soc., (2009), 131, 11949-11963; b) Hatakeyama T., Nakamura M., J. Am. Chem. Soc., (2007), 129, 9844-9845.
[160] Mayer M., Czaplik W.M., Jacobi von Wangelin A., Synlett, (2009), 2919- 2923.
[161] Ok H.O., Reigle L.B., Candelore M.R., Cascieri M.A., Colwell L.F., Deng L., Feeney W.P., Forrest M.J., Hom G.J., MacIntyre D.E., Stader C.D., Tota L., Wang P., Wyvratt M.J., Fisher M.H., Weber A.E., Bioorg. Med. Chem. Letters, (2000), 10, 1531-1534.
[162] Hatakeyama T., Kondo Y., Fujiwara Yu-J., Takaya H., Nakamura M., Ito Sh., Nakamura E., Chem. Commun., (2009), 1216-1218.
[163] Van Huis Ch.A., Casimiro-Garcia A., Bigge C.F., Cody W.L., Dudley D.A., Filipski K-J., Heemstra R.J., Kohrt J.T., Leadley Jr. R.J., Narasimhan L.S., McClanahan T., Mochalkin I., Pamment M., Thomas P.T., Sahasrabudhe V., Schaum R.P., Edmunds J.J., J. Bioorg. Med. Chem., (2009), 17, 2501-2511.
[164] Pat. WO 13126 (2009) (C.A., 2009, 150, 191522); Orsini P., Menichin-Cheri M., Vanotti E., Panzeri A., Tetrahedron Letters, (2009), 50, 3098-3100.
[165] Pat. WO 70869 (2009) (C.A., 2009, 151, 33423).
[166] Davies A.J., Scott J.P., Bishop B.C., Brands K.M., Brewer S.E., DaSilva J.O., Dormer P.G., Dolling U-H., Gibb A.D., Hammond D.C., Lieberman D.D., Palucki M., Payack J.T., J. Org. Chem., (2007), 72, 4864-4871.
[167] Pat. WO 140128 (2009) (C.A., 2009, 151, 571119).
[168] Abarbri M., Dehmel F., Knochel P., Tetrahedron Letters, (1999), 40, 7449- 7454.
[169]. ЗАО НПО «Пим-Инвест». Синтезы фторорганических соединений., Изд. «Тровант», Москва, (2005), c. 123, 124.
[170] Scheuermann G.M., Steurer P., Muelhaupt R., Rumi L., Bannwarth W., J. Am. Chem. Soc., (2009), 131, 8262-8270.
[171] Zhang X., Mu F., Robinson B., Wang P., Tetrahedron Letters, (2010), 51, 600-601.
[172] Audia J.E., Mergott D.J., Sheehan S.M., Watson B.M., Pat. US 275566 (2009) (C.A., 2009, 151, 528778).
[173] Pat. WO 103007 (2009) (C.A., 2009, 151, 288987).
[174] Gordeev M.F., Pat US 48305 (2009) (C.A., 2009, 150, 214368).
[175] Pulici M., Zuccotto F., Badari A., Nuvoloni S., Cervi G., Traquandi G., Biondaro S., Trifiro P., Marchionni C., Modugno M., Pat. WO 10154 (2010) (C.A., (2010), 152, 215278).
[176] Liu J., Jian T., Guo L., Atanasova T., Nargund R.P., Tetrahedron Letters, (2009), 50. 5228-5230.
[177] Heiss C., Schlosser M., Eur. J. Org. Chem., (2003), 447-451.
[178] Morgan B.P., Galdamez G.A., Gilliard Jr. R.J., Smith R.C., Dalton Trans., (2009), 2020-2028.
[179] a) Organ M.G., Calimsiz S., Sayah M., Hoi K.H., Lough A.J., Angew. Chem., Int. Ed.Engl., (2009), 48, 2383-2387; b) Altenhoft G., Goddard R., Lehmann Ch.W., Glorius F., J. Am. Chem. Soc., (2004), 126, 15195-15201.
[180] Sakamoto Y., Suzuki T., Miura A., Fujikawa H., Tokito S., Taga Y., J. Am. Chem. Soc., (2000), 122, 1832-1833.
[181] Yudin A.K., Martyn L.J.P., Pandiaraju S., Zheng J., Lough A., Org. Letters, (2000), 2, 41-44.
[182] Pat. WO 111299 (2009) (C.A., 2009, 151, 358919).
[183] Burton D.J., Hartgraves G.A., J. Fluor.Chem., (2009), 130, 254-258.
[184] Виноградов А.С., Краснов В.И., Платонов В.Е., Ж. орган. хим., (2008), 44, 101-107. Vinigradov A.S., Krasnov V.I., Platonov V.E., Russ. J. Org. Chem., (2008), 44, 95-102 (English translation).
[185] Hori A., Kataoka H., Akacaka A., Okano T., Fujita M., J. Polymer Sci., part A: Polymer Chem., (2003), 41, 3478-3485.
[186] Facchetti A., Yoon M-H., Stern Ch.L., Katz H.E., Marks T.J., Angew. Chem., Int. Ed. Engl., (2003), 42, 3900-3903. 114
[187] Cho D.M., Parkin S.R., Watson M.D., Org. Letters, (2005), 7, 1067-1068.
[188] Blass B.E., Huang C.T., Kawamoto R.M., Li M., Liu S., Portlock D.E., Rennells W.M., Simmons M., Bioorg. Med. Chem. Letters, (2000), 10, 1543-1546.
[189] Ragni R., Orselli E., Kottas G.S., Omar O.H., Babudri F., Pedone A., Naso F., Farinola G., DeCola L., Chemistry – A Europ. J., (2009), 15, 136-148.
[190] Nagao I., Shimizu M., Hiyama T., Angew. Chem., Int. Ed. Engl., (2009), 48, 7573-7576.
[191] Frattarelli D., Ratner M.A., Marks T.J., Schiavo M., Facchetti A., J. Am. Chem. Soc., (2009), 131, 12595-12612.
[192] Pat. WO 106577 (2009) (C.A., 2009, 151, 313566).
[193] Pat. EP 1674452 (2006) (C.A., 2005, 142, 355051); Pat. EP 1780197 (2007) (yield 83%) (C.A., 2006, 144, 191974); Tecle H., Shao J., Li Y., Kuthe M., Kazmirski S., Penzotti J., Ding Y-H, Moshinsky D., Coli R., Jhawar N., Bora E., JaquesO’ Hagan S., Wu J., Ohren J., Bioorg. Med. Chem. Letters, (2009), 19, 226-229 (yield 65%).
[194] Wang C.H., Alluri S., Ganguly A.K., Tetrahedron Letters, (2009), 50, 1879- 1881.
[195] Matsuda Sh., Takahashi M., Monguchi D., Mori A., Synlett, (2009), 1941- 1944.
[196] Huang Ch., Gevorgyan V., J. Am. Chem. Soc., (2009), 131, 10844-10845.
[197] Geramita K., McBee J., Tilley T.D., J. Org. Chem., (2009), 74, 820-829.
[198] Marchetti F., Pampaloni G., Passarelli V., Masi F., Sommazzi A., Spera S., J. Fluor. Chem., (2009), 130, 341-347.
[199] Pat. WO 117097 (2009) (C.A., 2009, 151, 403303).
[200] Baumann K., Flohr A., Goetschi E., Jacobsen H., Jolidon S., Luebbers T., Pat. US 181965 (2009) (C.A., 2009, 151, 148313).
[201] Oswald C.L., Peterson J.A., Lam H.W., Org. Letters, (2009), 11, 4504-4507.
[202] Mariaca R., Labat G., Behrnd N-R., Bonin M., Helbling F., Eggli P., Couderc G., Hulliger J., Neels A., Stoeckli-Evans H., J. Fluor. Chem., (2009), 130, 175-196.
[203] Jayanth T.T., Zhang L., Johnson T.S., Malinakova H.C., Org. Letters, (2009), 11, 815-818.
[204] Takayuki T., Yuhei Y., Masashi K., Toshiaki M., Org. Letters. (2009), 11, 1043-1045.
[205] Martin R.E., Wytko J.A., Diederich F., Helv. Chim. Acta, (1999), 82, 1470- 1479.
[206] Rosenblum S.B., Huynh T., Afonso A., Davis Jr. H.R., Tetrahedron, (2000), 56, 5735-5742.
[207] Pat. US 42841 (2009) (C.A., 2009, 150, 168179).
[208] Lledo A., Restorp P., Rebeck Jr. J., J. Am. Chem. Soc., (2009), 131, 2440- 2444.
[209] a) Isaac M., Slassi A., Silva K.D., Xin T., Tetrahedron Letters, (2001), 42, 2957-2960; b) Ying M., Smentek M.G., Day C.S., Welker M.E., Ma R., Torti S.V., Letters in Org. Chem., (2009), 6, 242-251.
[210] Matsnev A., Noritake S., Nomura Y., Tokunaga E., Nakamura S., Shibata N., Angew. Chem., Int. Ed. Engl., (2010), 49, 572-576.
[211]. Biffis A., Scattolin E., Ravasio N., Zaccheria F., Tetrahedron Letters, (2007), 48, 8761-8764; Luque R., MacQuarrie D.J., Org. Biomol. Chem., (2009), 7, 1627- 1632; Johnson S.A., Liu F-Q., Suh M.C., Zuercher S, Haufe M., Mao S.S.H., Tilley T.D., J. Am. Chem. Soc., (2003), 125, 4199-4211; Zeidan T.A., Kovalenko S.V., Monoharan M., Clark R.J., Ghiviriga J., Alabugin I.V., J. Am. Chem. Soc., (2005), 127, 4270-4285; Smith C.E., Smith P.S., Thomas R.L., Robins E.G., Collings J.C., Dai Ch., Scott A.J., Borwick S., Batsanov A.S., Watt S., Clark C.J., VineyCh., Howard J.A.K., Clegg W., Marder T.B., J. Mater. Chem., (2004), 14, 413-420.
[212] a) Ojima I. Fluorine in Medicinal Chemistry and Chemical Biology, J. Wiley Publication , Willy-Blackwell, (2009), p. 299, 301; b) Benmansour H., Chambers R.D., Sandford G., Yufit D.S., Hovard J.A.K., Arkivoc, (2007), (XI), 46-55.
[213] Amii H., Uneyama K., Chem. Revs., (2009), 109, 2119-2183.
[214] Deck P.A., Kroll C.E., Hollis W.G., Fronczek F.R., J. Organomet. Chem., (2001), 637-639, 107-115.
[215] Deck P.A., McCauley B., Slebodnick C., J. Organomet. Chem., (2006), 691, 1973-1983.
[216] Gurjar M., Reddy D.S., Murugaiah A., Murugaiah S., Synthesis, (2000), 1659- 1661.
[217] [212a, p. 307].
[218] Sasaki Sh., Tanabe Y., Yoshifuji M., Bull. Chem. Soc. Japan, (1999), 72, 563- 572.
[219] Morrison D.J., Trefz T.K., Piers W.E., McDonald R., Parvez M., J. Org. Chem., (2005), 70, 5309-5312.
[220] Yang Zh-Yu., Burton D.J., J. Fluor. Chem., (2000), 102, 89-103.
[221] Wang Y., Parkin S.R., Giersehner J., Watson M.D., Org. Letters, (2008), 10, 3307-3310.
[222] Wood T.K., Waren W.E., Keay B.A., Parvez M., Angew. Chem., Int. Ed. Engl., (2009), 48, 4009-4012.
[223] Nitschke J. K., Tilley T.D., J. Am. Chem. Soc., (2001), 123, 10183-10190.
[224] Zahn A., Brotschi C., Leumann C.J., Chemistry – A Europ. J., (2005), 11, 2125-2129.
[225] Pat. WO 93467 (2006) (C.A., 2006, 145, 326323).
[226] Ishihara K., Hasegawa A., Yamamoto H., Angew. Chem., Int. Ed. Engl., (2001), 40, 4077-4079.
[227] Crouch D.J., Skabata P.J., Heeney M., Culloch I., Coles S.J., Hursthouse M.B., Chem. Commun., (2005), 1465-1467.
[228] Wang Y., Parkin S.R., Watson M.D., Org. Letters, (2008), 10, 4421-4424.
[229] Deck P.A., Lane M.J., Montgomery J.K., Slebodnick C., Frouczek F.R., Organometallics, (2000), 19, 1013-1024.
[230] Caster K.C., Keck Ch. G., Walls R.D., J. Org. Chem., (2001), 66, 2932-2936.
[231] Schlosser M., Guio L., Leroux F., J. Am. Chem. Soc., (2001), 123, 3822- 3823.
[232] Coe J.W., Wirtz M.C., Bashore C.G., Candler J., Org. Letters, (2004), 6, 1589- 1592.
[233] Pat. WO 128142 (2006) (C.A., 2007, 146, 27723).
[234] Pat. US 6362365 (2002) (C.A., 2000, 132, 308136).
[235] Tang G., Yang Y.G., Wen J.X., Mol. Cryst. Liquid Cryst. Science and Technology. Sect. A: Mol. Cryst. and Liquid Cryst., (2002), 373, 17-24.
[236] Pat WO 40131 (2006) (C.A., 2006, 144, 390561).
[237] Nuritomi M., Murofushi H., Nakashima N., Bull. Chem. Soc. Japan, (2004), 77, 2121-2128.
[238] Dutta T., Woody K.B., Watson M.D., J. Am. Chem. Soc., (2008), 130, 452- 453.
[239] Yan Sh-J., Huang Ch., Zeng X-H., Huang R., Lin J., Bioorg Med. Chem. Letters, (2010), 20, 48-51. 117
[240] Mallach E., Kuhn N., Maichle-Moessmer C., Steimann M., Stroebele M., Zeller K-P., Z. Naturforsch. B. Chemical Sciences, (2009), 64, 1176-1182.
[241] Pat. US 5856557 (1999) (C.A., 1998, 128, 88679).
[242]. Bella M., Kobbelgaard S., Jørgensen K.A., J. Am. Chem. Soc., (2005), 127, 3670-3671.
[243] Kobbelgaard S., Bella M., Jørgensen K.A., J. Org. Chem., (2006), 71, 4980- 4987.
[244] Steffen A., Sladek M.J., Braun T., Neumann B., Stammler H.G., Organometallics, (2005), 24, 4057-4064.
[245] Schaub T., Backes M., Radius U., J. Am. Chem. Soc., (2006), 128, 15964- 15965.
[246] Braun T., Perutz R.N., Sladek M.I., Chem. Commun., (2001), 2254-2255.
[247] Yoshikai N., Mashima H., Nakamura E., J. Am. Chem. Soc., (2005), 127, 17978-17979.
[248] Wang J-R., Manabe K., Org. Letters, (2009), 11, 741-744.
[249] Ishikawa S., Manabe K., Synthesis, (2008), 3180-3182; Manabe K., Ishikawa S., Synthesis, (2008), 2645-2649.
[250] Braun T., Isundu J, Steffen A., Neumann B., Stammler H-G., Dalton Trans., (2006), 5118-5123.
[251] Guo H., Kong F., Kanno K., He J., Nakajima K., Takahashi T., Organometallics, (2006), 25, 2045-2048.
[252] Korn T.J., Schade M.A., Cheemala M.N., Wirth S., Guevata S.A., Cahiez G., Synthesis, (2006), 3547-3574; Korn T.J., Schade M.A., Wirth S., Knochel P., Org. Letters, (2006), 8, 725-728.
[253] Edelbach B.L., Kraft B.M., Jones W.D., J. Am. Chem. Soc., (1999), 121, 10327-10331.
[254] Miller A.O., Krasnov V.I., Peters D., Platonov V.E., Miethchen R., Tetrahedron Letters, (2000), 41, 3817-3819.
[255] Buckley H.L., Sun A.D., Love J.A., Organometallics, (2009), 28, 6622-6624; Wang T., Love J.A., Organometallics, (2008), 27, 3290-3296.
[256] Blay G., Fernandez I., Monleon A., Pedro J-R., Vila C., Org. Letters, (2009), 11, 441-444. 118
[257] Musso D.L., Cochran F.R., Kelley J.L., McLean Ed.W., Selph J.L., Rigdon G.C., Orr G. Faye, Davis R.G., Cooper B.R., Styles V.L., Thompson J.B., Hall W.R., J. Med. Chem., (2003), 46, 399-408.
[258] Tagat J.R., McCombic S.W., Nazareno D.V., Boyll C.D., Kozlowski J.A., Chackalamannil S., Josien H., Wang Ya., Zhou G., J. Org. Chem., (2002), 67, 1171- 1177.
[259] Andrus M.B., Liu J., Tetrahedron Letters, (2006), 47, 5811-5814; Moran B.N., Anderson F.P., Cloonan S., Butler N.E., Kenny P.T.M., Deverg A., Verughese S., Draper S.M., Bioorg. Med. Chem., (2009), 17, 4510-4522.
[260] Cornella J., Lu P., Larrosa I., Org. Letters, (2009), 11, 5506-5509.
[261] Shang R., Fu Y., Wang Y., Xu Q., Yu H-Z., Liu L., Angew. Chem., Int. Ed. Engl., (2009), 48, 9350-9354.
[262] Wyatt P., Hudsen A., Charmant J., Orpen A.G., Phetmung H., Org. Biomol. Chem., (2006), 4, 2218-2232.
[263] Iovel I, Mertins K., Kirschel J., Zapf A., Beller M., Angew. Chem., Int. Ed. Engl., (2005), 44, 3913-3917.
[264] Li L., Chen H., Lin Y., Synth. Commun., (2007), 37, 985-991.
Материал рекомендован к публикации членом редколлегии В.Е. Платоновым
Fluorine Notes, 2014, 93, 1-2


