Поступило в редакцию: февраль 2014
Fluorine Notes, 2014, 93, 7-8
Теоретическое исследование структурных свойств, энергий и природы орбитальных связей новых фторорганических соединений
Shahriar Ghammamy a,*, NoorAhmad Qaitmas a, Amir Lashgari a
Department of Chemistry, Faculty of Science, Imam Khomeini International University, Qazvin,
Iran
e-mail: ikiu2014@gmail.com, Tel: (+98)
281-8371378 Fax: (+98) 281-3780040
Аннотация: В представленном теоретическом исследовании мы использовали теорию функциональной плотности для вычисления молекулярной структуры соединения фторкарбоната, которым является COF2. Молекулярная геометрия, вибрационные частоты, энергии и естественные орбитали связи (NBO) в базовом состоянии вычисляются с использованием методов ТФП (B3LYP) с 3-21G. Вычисления производились на уровне ТФП (DFT), использовались разновалентные плюс поляризация 3-21G базисные наборы. The T.S guesses были генерированы с помощью линейного синхронного транзитного метода, на уровне ТФП DFT, внедренной в программу Gaussian 98. Местные минимумы были получены с помощью полной геометрической оптимизации, все они имеют положительные частоты. Эта молекула имеет Cs симметрии и форму дуги. B3LYP вычисленные вибрационные частоты масштабированы единичным фактором 0.88 и 0.96 для всех молекул, чтобы скорректировать известную систематическую 10-12% частоту по общей оценке. Инфракрасные интенсивности (int) в километрах на моль выполнялись на том же уровне на соответствующих полностью оптимизированных геометриях.
Ключевые слова: Теоретические исследования, DFT ТФП, B3LYP, NBO, COF2.
Введение
В органической химии существует три главных элемента: углерод, кислород и водород. В каждом органическом соединении содержится, по крайней мере, один атом углерода. Разграничение органических и неорганических соединений – всего лишь условность, и существует несколько соединений, которые классифицируются тем или иным образом, таких как: COCl2, CSCl2, CS(NH2)2, CO(NH2)2. Существует богатое разнообразие органической химии, которое не попадает в рамки органической химии и поэтому называется неорганической углеродной химией. Есть множество оксидов углерода (оксоуглеродов), из которых диоксид углерода (CO2) и монооксид углерода (CO) самые распространенные. Фторуглероды, иногда обозначаемые как перфторуглероды или PFCs – это фторные органические соединения, содержащие только углерод и фтор, связанные вместе в крепкие углеродно-фторные связи. Фторалканы, содержащие только одиночные связи, химически и термально более стабильны, нежели алканы. Однако, фторуглероды с двойной связью (фторалкены) и в особенности с тройной (fluoroalkynes) более реакционноспособны, чем соответствущие им углеводороды. Фторалканы можно использовать в качестве маслоотталкивающих/водоотталкивающих фторполимеров, растворителей и мощных парниковых газов. Существует тенденция использования ненасыщенных фторуглеродов как реагентов. Фторуглеродные жидкости являются бесцветными. У них большая плотность, до двух раз больше, чем у воды из-за их высокой молекулярной массы. Низкие внутримолекулярные значения энергии придают жидкостям низкую вязкость при сравнении с жидкостями с той же температурой кипения. Также заметны низкое значение поверхностного натяжения и показатели преломления. Они не смешиваются с большинством органических растворителей (т.е., этанолом, ацетоном, этилацетатом и хлороформом), но они смешиваются с некоторыми углеводородами (т.е. гексаном в некоторых случаях). Они плохо растворяются в воде, и растворимость их в воде очень низка (порядка 10 частей на миллион). Число атомов углерода в молекуле фторуглерода в большинстве своем определяет почти все физические свойства. Чем выше число атомов углерода, тем выше температура кипения, плотность, вязкость, поверхностное натяжение, главные свойства, давление пара и показатель преломления. Растворимость газа снижает рост атомов углерода, в то время как температура плавления определяется другими факторами и поэтому не предсказывается с готовностью. Множество различных данных по структурным свойствам соединений фторкарбонатов было открыто, но они недостаточны и противоречивы в некоторых аспектах. COF2 - это примитивное синтезированное вещество, которое используется для структурных химических исследований и органического синтеза [1-7].
Теоретические расчеты использовались для выявления структурных и электронных данных многих соединений, особенно, соединений фторкарбоната. В данном исследовании применялись методы теории функциональной плотности для определения оптимизированной структуры COF2. Никаких теоретических расчетов не было найдено для данного типа соединений, в особенности вычислений теории функциональной плотности (DFT). Вычисления производились при использовании программ Гасян 98 для ТФП, использовался трех-параметрный обменный функционал Бека в сочетании с функционалом корреляции Ли-Янг-Парра с 3-21G базисным набором [8-10]. Seppelt синтезировал новое соединение фторкарбоната, и в данной статье мы изучаем его свойства.
Методы и материал
Методы расчета
Все расчеты выполнены с помощью программы Gaussian 98, сочетающей в себе точный обмен Хартри-Фока с Беком и использующий функцию корреляции Ли-Янг-Парра для того, чтобы включать самые важные эффекты корреляции. Строение молекулы было полностью оптимизировано без какой-либо симметрии на всех уровнях. Оптимизированные структурные параметры использовались для вычисления вибрационной частоты на уровнях DFT для описания/ характеризации всех стационарных точек как минимальных. Интенсивности инфракрасного излучения (int), измеряемые в километрах на моль соединения выполнялись на том же уровне на соответственных полностью оптимизированных геометрических характеристиках. Данное соединение и его данные согласуются с недавними работами по образованию четырех полупродуктов координат.
Результаты
Соединение фторкарбоната, COF2 было исследовано, и были проведены оптимизации размеров/геометрии на уровне the DFT/ 3-21G level, они показаны на Рис. 1. COF2, в котором C (1) связан с атомом O (2), имеет линейную структуру C-O с длиной связи равной 1.19 Å. C(1) связан с атомом F(3) и атомом (4), обладает линейной структурой C–F с длиной связи равной 1.35 Å имеет изогнутую O– C –Fструктуру с углом связи равным 126. Выбранные расстояния связей показаны на Рис. 1. Геометрические характеристики соединения оптимизированы на уровнях B3LYP/ 3-21G. Использовались методы теории функциональной плотности для определения оптимизированной структуры COF2. Первоначальные вычисления выполнялись на уровне DFT, использовались также level and split- valence plus polarization 3-21G basis sets were used. Локальные минимумы были получены с помощью полной геометрической оптимизации, все они с положительными частотами. Все вычисления выполнялись с помощью компьютерной программы GAUSSIAN 98.
И высшая занимаемая молекулярная орбиталь (HOMO) и низшая незанимаемая молекулярная орбиталь (LUMO) являются главными орбиталями, что принимают участие в химической устойчивости. HOMO представляет собой способность производить электрон, LUMO в качестве акцептора электрона представляет собой способность получать электрон, энергии HOMO и LUMO вычисленные с помощью B3LYP на 3-21G метода представлены на Рисунке 2. Данная абсорбция электронов соответствует переходу из базового к первому возбужденному состоянию и главном образом, описывается одним электронным возбуждением с наивысшей занимаемой орбитали (LUMO).
Атомные заряды и кратности связи являются существенными параметрами для нашего исследования. Данные количества получены из анализа популяции NBO. Метод NBO является предпочтительным для зарядов Mulliken, так как последний обеспечивает картину орбитали, которая ближе к классической структуре Льюиса. Анализ NBO, затрагивающий атомные заряды, кратности связей, а также гибридизации избранных связей вычислены на уровне B3LYP/ 3-21G. Гибридизация данной молекулы это SP, что подтверждается структурой Таблицы 2. Анализ теории пертрубации\возмущения второго порядка матрицы Фока на основе NBO для COF2 показан в таблице 3. У этой молекулы симметрии Cs и дуговая форма.
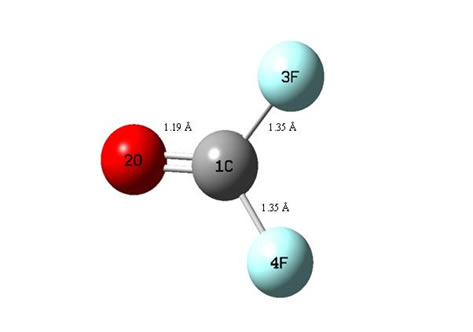
Рисунок 1.Оптимизированные геометрии COF2 на B3LYP/BS1 уровне теории.
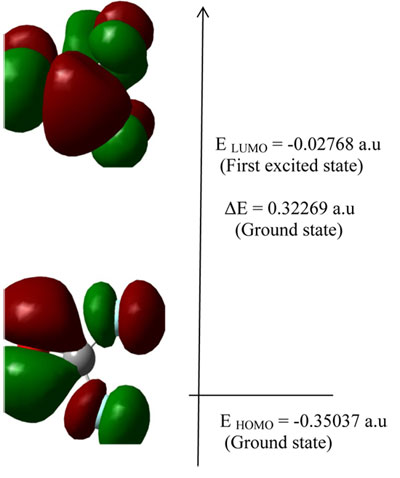
Рисунок 2. Композиции атомной орбитали передней\пограничной молекулярной орбитали COF2.
Таблица 1. Сравнение вычисляемых углов связи (в градусах) для вещества.
|
Вещество |
Θ |
|
COF2 |
ΘO2C1F3=126 |
|
COF2 |
ΘO2CF4=126 |
|
COF2 |
ΘF3CF4=107.8 |
Таблица 2. The NBO вычисляемые гибридизации COF2.
|
Связь |
Атом |
COF2 |
|
C-O(2) |
C |
S1P1.49 |
|
O(2) |
S1P1.61 |
|
|
C-O(2) |
C |
S1P1 |
|
O(2) |
S1P1 |
|
|
C-F(3) |
C |
S1P2.34 |
|
F(3) |
S1P3.26 |
|
|
C-F(4) |
C |
S1P2.34 |
|
F(4) |
S1P3.26 |
|
|
- |
O(2) |
S1P0.62 |
|
- |
O(2) |
S1P1 |
|
- |
F(3) |
S1P0.31 |
|
- |
F(3) |
S1P99.99 |
|
- |
F(3) |
S1P1 |
Таблица 3. Анализ теории пертрубации\возмущения второго порядка матрицы Фока на основе NBO для COF2. a E(2) означает энергию гиперсопряженного взаимодействия (энергия стабилизации); b Разница энергии между донорной и акцепторной i и j NBO орбиталями; c F(i, j) – это элемент матрицы Фока между i и j NBO орбиталями.
|
Донор (i) |
Тип |
ED/e |
Акцептор (j) |
Тип |
ED/e |
E(2) a(KJ/mol) |
E(j)- E(i) b(a.u) |
F(i,j)c (a.u) |
|
CO2 (2) |
σ |
1.99976 |
CO2 (2) |
σ* |
0.25720 |
2.61 |
0.37 |
0.030 |
|
CF3 (1) |
σ |
1.99049 |
CF4 (1) |
σ* |
0.16333 |
3.22 |
1.20 |
0.058 |
|
CF4 (1) |
σ |
1.99049 |
CF3 (1) |
σ* |
0.16333 |
3.22 |
1.20 |
0.058 |
|
O2 (1) |
n |
1.97365 |
CF3 (1) |
σ* |
0.16333 |
1.71 |
0.92 |
0.037 |
|
O2 (1) |
n |
1.97365 |
CF4 (1) |
σ* |
0.16333 |
1.71 |
0.92 |
0.037 |
|
F3 (1) |
n |
1.98849 |
CO2 (1) |
σ* |
0.04069 |
1.03 |
1.68 |
0.037 |
|
F4 (1) |
n |
1.98849 |
CO2 (1) |
σ* |
0.04069 |
1.03 |
1.68 |
0.037 |
|
CF3 (1) |
σ* |
0.16333 |
CO2 (1) |
σ* |
0.04069 |
1.46 |
0.37 |
0.065 |
|
CF4 (1) |
σ* |
0.16333 |
CO2 (1) |
σ* |
0.04069 |
1.46 |
0.37 |
0.065 |
Обсуждение
Вычисления производились с помощью программы Gaussian 98 и программы визуализации молекулы Gauss View 5.0. Структуры молекул были полностью оптимизированы без симметрии на всех уровнях. Оптимизированные структурные параметры использовались в вычислениях вибрационных частот на уровнях DFT чтобы характеризовать все стационарные точки как минимумы. Оптимизации геометрии вычислений DFT производились с помощью трех-параметрового гибридного функционала Бека с функцией корреляции Ли-Янг-Парр, комбинацией которая дает подъем хорошо известному методу B3LYP. Базисный набор 3-21G использовался для всех методов молекул B3LYP. Harmonic vibrational frequencies were computed using B3LYP methods with the 3-21G basis sets. The anharmonicity of the fundamental frequencies is most often taken into account by scaling the calculated harmonic frequencies, and this procedure has been found to work well since the overestimation of vibrational frequencies is fairly uniform. The B3LYP calculated vibrational frequencies have been scaled by a single factor of 0.88 and 0.96 for all molecules to correct the well-known systematic 10–12% frequency over estimation. Infrared intensities (int) in Kilometer per mole of compound were performed at the same level on the respective fully optimized geometries. This compound and their data are in accordance with recent works on the formation of four coordinate intermediates.
Благодарность
Выражаем глубокую признательность и благодарность за финансовую поддержку Совету по исследованиям Международного Университета Имама Хомейни.
Список литературы
- Lemal, D.M., 2004. Perspective on fluorocarbon chemistry. J. Org. Chem., 69: 1–11.
- Lewandowski, G., E. Meissner, E. Milchert, 2006. Special applications of fluorinated organic compounds. J. Hazard. Mater, 136: 385–91.
- O'Hagan, D., 2008. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev., 37: 308–19.
- Kiplinger J.L., Richmond TG, Osterberg CE (1994). "Activation of Carbon-Fluorine Bonds by Metal Complexes". Chem. Rev. 94 (2): 373–431.
- Murphy C.D., C. Schaffrath, D., 2003. O'Hagan: Fluorinated natural products: the biosynthesis of fluoroacetate and 4-fluorothreonine in Streptomyces cattleya. Chemosphere, 52:455-61.
- Steenland, K., T. Fletcher, D.A. Savitz, 2010. Epidemiologic Evidence on the Health Effects of Perfluorooctanoic Acid (PFOA) . Environmental Health Perspectives, 118: 1100–8.
- Berthelot, M., 1891. Action de la chaleur sur l'oxyde de carbone. Annales de Chimie et de Physique, 6: 126–132.
- Ando, Sh., 2006. DFT Calculations on Refractive Index Dispersion of Fluoro-compounds in the DUV-UV-Visible Region. Journal of Photopolymer Science and Technology, 19: 351-360.
- Audi, G., 2003. The Nubase Evaluation of Nuclear and Decay Properties. Nuclear Physics,729: 3–128.
- Becke, A.D., 1993. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys., 98: 5648-5652.
Статья рекомендована к публикации членом редколлегии Д.х.н. С.М. Игумновым
Fluorine Notes, 2014, 93, 7-8


