Поступило в редакцию: декабрь 2013
Fluorine Notes, 2014, 93, 3-4
Новый путь синтеза γ-карболинов и 5-азаиндолов с использованием 3,3,3-трифтор-1-нитропропена
Федоровский О.Ю.а, Гусев Д.В.а, Чкаников Н.Д.а, Иванов А.В.б, Щербакова В.С.б
а Федеральное государственное бюджетное учреждение науки Институт элементоорганических
соединений им. А.Н.Несмеянова Российской академии наук, 119991, ГСП-1, Москва, В-334, ул.
Вавилова, д. 28
e-mail: offskii@rambler.ru
а Иркутский институт химии им. А. Е. Фаворского СО РАН, ул. Фаворского, 1, 664033,
Иркутск, Россия
e-mail: ivanov@irioch.irk.ru
Аннотация. Получен ряд новых фторсодержащих производных γ-карболинов и 5-азаиндолов с использованием 3,3,3-трифтор-1-нитропропена.
Ключевые слова: Фторсодержащие γ-карболины, фторсодержащие 5-азаиндолы, 3,3,3-трифтор-1-нитропропен, 4,5,6,7-тетрагидроиндол, циклизация Пикте-Шпенглера.
Ранее нами был предложен удобный метод синтеза конденсированных 3-трифторметилпиридинов с использованием 3,3,3-трифтор-1-нитропропена (2). Так, исходя из индола и алкена (2) получены β-карболины. Ключевыми промежуточными соединениями были производные триптамина, полученные восстановлением продуктов С3-алкилирования индола алкеном (2) [1]. Недавно нами показано, что производные 4,5,6,7-тетрагидроиндола (ТГИ) (1a), ставшего в последние годы легкодоступным замещенным пирролом [2], региоселективно и количественно оксиалкилируются полифторкарбонильными соединениями по С2 углеродному атому пиррольного кольца [3]. Такой результат позволил надеяться, что С2-алкилирование ТГИ алкеном (2) откроет путь к производным изо-триптамина и далее к фторсодержащим γ-карболинам, представляющим большой интерес как потенциальные лекарственные препараты [4, 5, 6]. В настоящей работе показано, что такая синтетическая схема, действительно, приводит к получению насыщенных производных дигидро-, тетрагидро-, и октагидро-γ-карболинов, предшественников ароматических γ-карболинов (см. схема 2). Последние могут быть получены дегидрированием насыщенных γ-карболинов и удалением защитных групп известными методами [7]. Эта же схема превращений использовалась в данной работе и для синтеза фторсодержащих 5-азаиндолов (см. схема 3), представляющих безусловный интерес при поиске новых лекарственных средств [8, 9]. В соответствии с авторами [6] мы использовали общепринятую нумерацию для полученных соединений (см. схема 2).
Обсуждение результатов
При взаимодействии ТГИ в растворе абсолютного эфира с 3,3,3-трифтор-1-нитропропеном (2) образовывался неустойчивый продукт реакции, который за несколько часов разлагался даже при пониженной температуре (от -70 до 0°С) в атмосфере аргона. Замена ТГИ на N-винил-ТГИ в реакции с алкеном (2) стабильность нитроаддукта (3) не увеличивает.
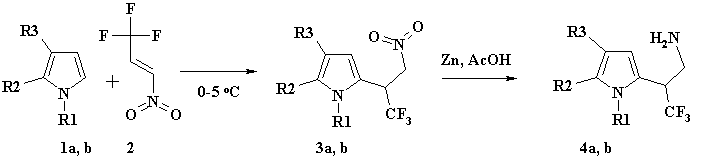
Схема 1. (a: R1=Bnz, R2-R3= -(CH2)4-; b: R1= -Me, R2=R3= H)
Продукт реакции (3a) оказался стабильным в случае защищённого ТГИ по атому азота бензильной группой. Так, N-Bnz-ТГИ (1a) гладко реагировал с 1,1,1-трифтор-3-нитропропеном (2) по второму положению пиррольного кольца в абсолютном эфире при 0 °С в атмосфере аргона с количественным выходом. Полученное нитропроизводное (3a) восстанавливалось до N-Bnz-изо-триптамина (4a) активированной цинковой пылью в растворе уксусной кислоты (см. схема 1). Далее, N-Bnz-изо-триптамин (4a) был введен в циклизацию с р-фенил-3-трифторметил-бензальдегидом в условиях реакции Пикте-Шпенглера [10] (см. схема 2).
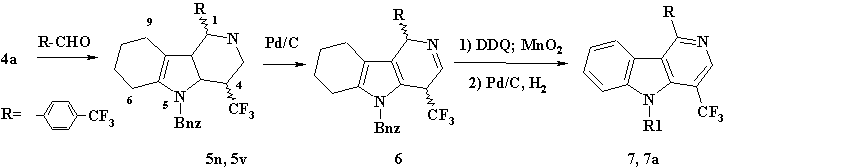
Схема 2. ((7) R1= Bnz; (7а) R1=H).
В результате этой реакции было получено 2 диастереомера (5n) и (5v) в соотношении 6/4 соответственно, которые были разделены хроматографически на силикагеле. Суммарный выход (5n) и (5v) составил 48%. Следует отметить, что при циклизации в растворе уксусной кислоты в присутствии каталитических количеств р-толуолсульфокислоты образуется только один диастереомер γ-карболина (5v) с выходом 50%.
В условиях каталитического дегидрирования, диастереомеры (5n) и (5v) имеют различную реакционную способность, что видимо, объясняется различным пространственным строением пиридинового кольца. Так, при кипячении в растворе бензола в присутствии 10% Pd/C диастереомер (5n) дегидрировался с образованием двойной связи в пиридиновом кольце, соединение (6). Соединение (5v) было устойчиво к дегидрированию в этих условиях. Длительное кипячение (5n, 5v, 6) в 1,3,5-триметилбензоле в присутствии DDQ приводило к образованию полностью ароматическое соединения (7) с низким выходом (см. схема 2). Использование активированного диоксида марганца для дегидрирования (5n) и (5v) в растворе 1,3,5-триметилбензола приводило к образованию соединения (7) с выходом 67%. Бензильная защитная группа была удалена кипячением (7) в растворе этанола в присутствии 10% Pd/C в атмосфере водорода с образованием (7а) с выходом 65%.
Безусловный интерес при поиске новых лекарственных средств, представляют также и 5-азаиндолы [11, 12], которые были получены нами в данной работе. В синтезе фторсодержащих 5-азаиндолов, использовалась схема превращений аналогичная получению γ-карболинов, исходя из N-метилпиррола (1b) и алкена (2) (см. схема 1).
Как в случае N-Bnz-ТГИ так и N-метилпиррола реакция при О°С протекала региоселективно по С2 углеродному атома пиррольного кольца с образованием нитрофторпроизводного (3b) с выходом близким к количественному, однако авторы [13] в этом же случае регеоселективности не наблюдали. Нитросоединение (3b) было восстановлено в 3,3,3-трифтор-2-(1-метил-1Н-пиррол-2-ил)-пропиламин (4b) активированным цинком в водном растворе уксусной кислоты с хорошим выходом (83%). Последнее, вводилось в циклизацию с альдегидами в мягких условиях (~50°С) по реакции Пикте-Шпенглера (см. схема 3) с образованием производных 4,5,6,7-тетрагидро-пирролопиридинов (8, 9). Эти соединения были получены в виде смеси диастереомеров. После очистки и разделения с помощью препаративной хроматографии на силикагеле, были выделены основные диастереомеры с выходами 65% и 55% для соединений (8) и (9) соответственно.
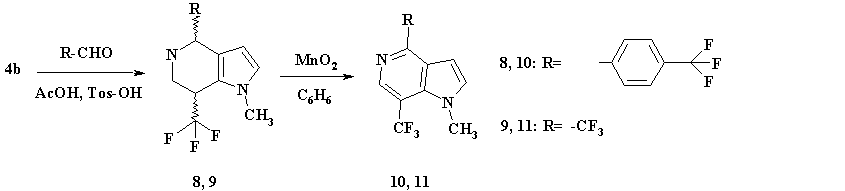
Дегидрирование соединений (8) и (9) проводилось в присутствии активированного диоксида марганца в растворе кипящего бензола с образованием производных 5-азаиндола (10, 11) с высокими выходами (см. схема 3).
Таким образом, по предлагаемой нами схеме синтеза аннелированных пиридинов с использованием 3,3,3-трифтор-1-нитропропена (2) могут быть получены фторсодержащие производные насыщенных и ароматических γ-карболинов и 5-азаиндолов.
Экспериментальная часть
Чистота и структуры полученных соединений подтверждены методами тонкослойной хроматографии, пластины Kieselgel 60 F254, ЯМР-спектроскопии Avance 300, для ядер 1H, 19F, и хроматомасс-спектрометрии Finnigan Polaris Q/Trace GC ultra, ионная ловушка, ЭУ 70Эв. Колонка: RTX-5ms, тип фазы: 5% phenylpolysilphenylene-siloxane.
1-Бензил-2-(2,2,2-трифтор-1-нитрометил-этил)-4,5,6,7-тетрагидро-1Н-индол (3а).
К 100 мл эфирного раствора, содержащего 7.1г (0.0336моль) N-Bnz-THI (1а) добавили 4.74г (0.0336моль) 1-нитро-3,3,3-трифторпропена (2) при 0-5 °С. Контроль за ходом реакции проводили методом ТСХ, элюент: хлороформ/этилацетат в соотношении 95/5. После завершения реакции (около 3 часов) эфирный раствор был упарен на роторном испарителе при пониженном давлении. Соединение (3а) было получено в количестве 11.8г практически с количественным выходом и использовалось в дальнейших превращениях без дополнительной очистки. Светло-жёлтое масло, Rf= 0.75.
Спектр ЯМР-1Н, dmso-d6: 1.6ppm, 4H, m, 5, 6 -CH2-; 2.2-2.4ppm, 4H, m, 4, 7 -CH2-; 4.6-4.7ppm, 1H, m, *CH-CF3; 4.9-5.3ppm, 4H, Ph-CH2-, N-CH2-; 6.11ppm, 1H, s, C3H; 6.9-7.3ppm, 5H, m, Bnz.
Спектр ЯМР-19F, dmso-d6: 9.8ppm, d, j=10Hz, -CF3 .
Масс-спектр: m/z 352 [M]+ (19).
2-(1-Бензил-4,5,6,7-тетрагидро-1Н-индол-2-ил)-3,3,3-трифтор-пропиламин (4а).
11г (0.031моль) нитросоединения (3а) растворили в 70мл 50% водной уксусной кислоты и добавили 10г активированного цинка небольшими порциями в течение 15 минут. Реакцию проводили в интервале температур от 40 до 50 °С и интенсивном перемешивании. Контроль за прохождением реакции проводили методом ТСХ, до исчезновении исходного (3а). Затем, реакционная масса было охлаждена до комнатной температуры и разбавлена эфиром (300 мл). Эфирную вытяжку отделили от водного раствора соли ацетата цинка и подщелачивали насыщенным водным раствором поташа, с концентрацией ~70%. После достижения значения рН 10 водного солевого раствора под слоем эфира, эфирный раствор, содержащий N-Bnz-изо-триптамин (4а) был упарен и продукт сырец был очищен на колонке с силикагелем, элюент: дихлорметан/метанол = 95/5. Получено 7г соединения (4а) с выходом 70%. Светло-жёлтое масло, Rf= 0.5.
Спектр ЯМР-1Н, dmso-d6: 1.5-1.8ppm, 4H, m, 5, 6-CH2-; 2.2-2.5ppm, 4H, m, 4, 7-CH2-; 2.7-3.1ppm, 2H, m, N-CH2-; 3.2-3.4ppm, 3H, s уширен, -NH2; 3.5-3.6ppm, 1H, m, *CH; 5.1ppm, 2H, d, J=7Hz, Bnz-CH2-; 5.9ppm, 1H, s, C3H, 6.9-7.3ppm, 5H, m, Bnz.
Спектр ЯМР-19F, dmso-d6: 9.6ppm, d, j=10Hz, -CF3 .
Масс-спектр: m/z 322 [M+] (23).
5-Бензил-4-трифторметил-1-(4-трифторметил-фенил)-2,3,4,5,6,7,8,9-октагидро-1Н-пиридо[4,3-b]индол (5).
К 25 мл раствора этилового спирта, содержащего 2.656г (0.0082 моль) N-Bnz-изо-триптамина (4а) добавили 1.43г (0.0088 моль) 4-трифтор-метилбензальдегида и 0.5мл концентрированной соляной кислоты. Реакционную массу кипятили в течение 3 часов при перемешивании и с обратным холодильником. После завершения реакции (контроль по ТСХ) избыток соляной кислоты был нейтрализован добавлением 2г поташа, реакционная масса была отфильтрована и упарена, органическая масса, содержащая смесь двух диастереомеров 1,2,3,4,6,7,8,9-октагидро-γ-карболина (5n, 5v) была очищена хроматографически на силикагеле, с разделением диастереомеров. Элюент: дихлорметан/петролейный эфир в соотношении 9/1. Соединение (5n) было выделено в количестве 1.2г, соединение (5v) в количестве 0.7г. Общий выход γ-карболина (5) с учетом обоих диастереомеров составил 1.9г, 48%.
Соединение (5n): ТСХ: элюент хлороформ/этилацетат в соотношении 97/3, Rf= 0.3.
Спектр ЯМР-1Н, dmso-d6: 1.3-1.6ppm, 4H, m, 7, 8 -CH2-; 2.1-2.3ppm, 4H, m, 6, 9 –CH2-; 2.9-3.1ppm, 2H, m, C3H2; 3.4ppm, 1H, d, J=11Hz, C1H; 3.6ppm, 1H, m, C4H; 4.95ppm, 1H, s, уширен, NH; 5.05ppm, 2H, d, J=7Hz, Bnz-CH2-; 6.9-7.3ppm, 5H, m, Bnz, 7.5-7.7ppm, 4H, 2d, J=7Hz, p-Ph.
Спектр ЯМР-19F, dmso-d6: 13.1ppm, 3F, d, j=10Hz, -CF3; 17.6ppm, 3F, s, Ph-CF3.
Масс-спектр: m/z 478 [M]+ (100); 407 [M-CF3]+ (91), 380 [M-Bnz]+.
Соединение (5v): ТСХ: элюент хлороформ/этилацетат в соотношении 97/3, Rf= 0.7.
Спектр ЯМР-1Н, dmso-d6: 1.4-1.6ppm, 4H, m, 7, 8 -CH2-; 2.1-2.3ppm, 4H, m, 6, 9 -CH2-; 2.7ppm, 2H, m, C3H2; 3.1ppm, 1H, d, J=12Hz, C1H; 3.45ppm, 1H, m, C4H; 4.95ppm, 1H, s, уширен, NH; 5.1ppm, 2H, d, J=10Hz, Bnz-CH2-; 6.8, 7.6ppm, 4H, d, J=10Hz, p-Ph; 7.2-7.4ppm, 5H, m, Bnz.
Спектр ЯМР-19F, dmso-d6: 13.7ppm, 3F, d, j=10Hz, -CF3; 17.2ppm, 3F, s, Ph-CF3.
Масс-спектр: m/z 478 [M]+ (65); 387 [M-Bnz]+ (71); 380 [M-Bnz-7H]+ (81).
5-Безил-4-трифторметил-1-(4-трифторметил-фенил)-4,5,6,7,8,9-гексагидро-1Н-пирридо[4,3-b]индол (6).
К раствору 80мг (0.000167моль) соединения (5n) в 10мл бензола добавили 30 мг 10% Pd/C и реакционную массу перемешивали и кипятили с обратным холодильником 3 часа. Контроль за прохождением реакции проводили методом ТСХ, элюент: хлороформ/этилацетат в соотношении 95/5. Растворитель упарили на роторном испарителе при пониженном давлении и остаток очистили хроматографически на силикагеле, элюент: хлороформ/этилацетат в соотношении 95/5. Было получено 50мг соединения (6) с выходом 63%.
Спектр ЯМР-1Н, dmso-d6: 1.5-1.7ppm, m, 4H, 7,8 –CH2-; 2.0-2.2ppm, m, 6, 9 -CH2-; 3.7ppm, 1H, dd, J=10Hz, C3H; 4.05ppm, 1H, m, C4H; 4.45ppm, 1H, d, J=20Hz, C1H; 5.21ppm, 2H, s, Bnz-CH2-; 6.95-7.3ppm, 5H, m, Bnz; 7.6-7.8ppm, 4H, 2d, J=10Hz, p-Ph.
Спектр ЯМР-19F, dmso-d6: 13.1ppm, 3F, d, j=10Hz, -CF3; 17.3ppm, 3F, s, Ph-CF3.
Масс-спектр: m/z 476 [M]+ (6), 474 [M-2H]+ (14), 470 [M-6H]+ (18).
5-Бензил-4-трифторметил-1-(4-трифторметил-фенил)-5Н-пиридо[4,3-b]индол (7).
Процедура (а). К 10мл раствора 1,3,5-триметилбензола, содержащего 20мг (4.2*10-5 моль) (5n) добавили 60мг DDQ и кипятили трое суток. После исчезновения исходного карболина раствор упарили и остаток очистили на силикагеле, элюент: дихлорметан/петролейный эфир (49/70) в соотношении 8/2. Выделено ~5мг (7), выход 25%.
Процедура (b). Кипятили смесь, состоящую из 75мг (0.000157моль) (5n) и 75мг (0.000157моль) (5v) в присутствии 300мг активированного диоксида марганца в 15мл 1,3,5-триметилбензола в течение 3х часов и интенсивном перемешивании до исчезновения исходного (5), контроль за ходом реакции проводили методом ТСХ. Далее, аналогично процедуре (а). Выделено 100мг соединения (7), выход 67%.
Спектр ЯМР-1Н, dmso-d6: 5.8ppm, 2H, s, Bnz-CH2-; 6.9-7.6ppm, 9H, m, Bnz-, H6, H7, H8, H9; 7.8-8.00ppm, 4H, 2d, J=10Hz, p-Ph-; 8.95ppm, 1H, s, H3.
Спектр ЯМР-19F, dmso-d6: 15.1ppm, 3F, s, -CF3; 22.6ppm, 3F, s, -CF3.
Масс-спектр: m/z 470 [M]+ (36), 91 [Bnz]+ (100).
4-трифторметил-1-(4-трифторметил-фенил)-5Н-пиридо[4,3-b]индол (7а).
К раствору 10мл этанола, содержащего 150мг (3.2*10-4 моль) соединения (7) добавили 100мг 10% Pd/C, 100мг 36% НСl и подключили водородную камеру с давлением ~0.5 атм. Интенсивно перемешивали реакционную массу и нагревали до 50°С в течение суток. Контроль за ходом реакции проводили методом ТСХ до исчезновения исходного (7). Далее, реакционную массу профильтровали на целите и раствор упарили на роторном испарителе, добавили 30мл воды и 3г бикарбоната натрия. Экстрагировали эфиром (3 раза по 30мл), эфирные вытяжки объединили, сушили безводным сульфатом натрия и упарили при пониженном давлении. Чистили продукт на 50г силикагеля, элюент: дихлорметан/петролейный эфир в соотношении 8/2, Rf= 0.6. Выделили 80мг соединения (7а), выход 65%.
Спектр ЯМР-1Н, dmso-d6: 7.1-7.7ppm, 4H, m, C6H-C9H; 7.8-8.0ppm, 4H, 2d, J=10Hz, p-Ph; 8.37ppm, 1H, s уширен, NH, 8.67ppm, 1H, s, C3H.
Спектр ЯМР-19F, dmso-d6: 16.27ppm, 3F, s, -CF3; 17.94ppm, 3F, s, -CF3.
Масс-спектр: m/z 380 [M]+ (69), 379 [M-H]+ (100), 359 [M-HF]+ (27).
3,3,3-Трифтор-2-(1-метил-1Н-пиррол-2-ил)-пропиламин (4b).
К 25мл раствора 50% водной уксусной кислоты, содержащей 3.45г (0.0155моль) (3b) порционно добавили 3г активированного цинка в течение 5 минут. Нагревали реакционную массу до 40-50 °С при интенсивном перемешивании в течение 3 часов. Выделение и очистка (4а) идентична как для соединения (4а). Получено 2.4г (4b), выход 83%. Светло-жёлтое масло, Rf=0.2, элюент хлороформ/этилацетат в соотношении 9/1.
Спектр ЯМР-1Н, dmso-d6: 2.3ppm, 2H, s, уширен, -NH2; 3.1-3.4ppm, 2H, m, -CH2-; 3.55-3.55ppm, 1H, m, *CH; 3.61ppm, 3H, s, N-Me; 6.13-6.17ppm, 2H, m, CH, 6.63ppm, 1H, s, CH.
Спектр ЯМР-19F, dmso-d6: 8.2ppm, d, J=8Hz, -CF3.
Масс-спектр: m/z 192 [M]+ (37).
1-Метил-7-трифторметил-4-(4-трифторметил-фенил)-4,5,6,7-тетрагидро-1Н-пирроло[3,2-c]пиридин (8).
К раствору 10мл уксусной кислоты, содержащей 0.384г (0.002моль) (4b) добавили с 0.35г (0.002моль) р-фенил-3-трифторметилбензальдегида и каталитическое количество р-толуол-сульфокислоты (10мг). Реакционную массу нагревали при 50°С в течение двух часов. Затем, реакционная масса была упарена на роторном испарителе при пониженном давлении и перенесена в 25мл воды, содержащей 3г поташа. Водный слой экстрагировали эфиром 3 раза по 20мл, эфирные вытяжки объединили и сушили безводным сульфатом натрия. Очисткой на силикагеле, элюент хлороформ/этилацетат в соотношении 9/1 был выделен основной диастереомер (8) в количестве 0.45г с выходом 65%. Медленно кристаллизующееся масло, Rf=0.3.
Спектр ЯМР-1Н, dmso-d6: 2.7-2.9ppm, 1H, m, C6H; 3.0-3.15ppm, 1H, m, C6H; 3.4-3.5ppm, 1H, d, J=15Hz, C4H; 3.54ppm, 3H, s, N-Me; 3.7-3.8ppm, 1H, m, C7H; 4.92ppm, 1H, s, NH; 5.46ppm, 1H, d, J=3Hz, C3H; 6.65ppm, 1H, d, J=3Hz, C2H; 7.5-7.7ppm, 4H, 2d, J=8Hz.
Спектр ЯМР-19F, dmso-d6: 13.8ppm, 3F, d, J=13Hz, C7-CF3; 17.6ppm, 3F, s, p-Ph-CF3.
Масс-спектр: m/z 348 [M]+ (27).
1-Метил-4,7-вис-трифторметил-4,5,6,7-тетрагидро-1Н-пирроло[3,2-c]пиридин (9).
Получали аналогично (8). Выделено 0.3г (9) с выходом 55%. Медленно кристаллизующееся масло, Rf=0.7, элюент хлороформ/этилацетат в соотношении 9/1.
Спектр ЯМР-1Н, dmso-d6: 2.9-3.1ppm, 2H, m, C6H2; 3.3-3.4ppm, 1H, m, C4H; 3.54ppm, 3H, s, N-Me; 3.75-3.85ppm, 1H, m, C7H; 4.45-4.55ppm, 1H, m, уширен, NH; 5.95ppm, 1H, s, уширен, C3H; 6.8ppm, 1H, d, J=3Hz, C2H.
Спектр ЯМР-19F, dmso-d6: -64.5ppm, 3F, d, J=23Hz, -CF3; -75.5ppm, 3F, d, J=23Hz , -CF3.
Масс-спектр: m/z 272 [M]+ (34).
1-Метил-7-трифторметил-4-(4-трифторметил-фенил)-1Н-пирроло[3,2-c]пиридин (10).
К 10мл раствора бензола, содержащего 30мг (8.62*10-5моль) (8) добавили 50мг активированной двуокиси марганца. Реакционную массу при интенсивном перемешивании с обратным холодильником кипятили в течение двух часов. Контроль за ходом реакции проводили методом ТСХ. Бензол упарили на роторном испарителе и остаток чистили хроматографически на силикагеле, элюент: хлороформ/этилацетат в соотношении 9/1. После очистки выделено 25мг (10) с выходом 85%. Медленно кристаллизующееся масло, Rf=0.9, элюент хлороформ/этилацетат в соотношении 9/1.
Спектр ЯМР-1Н, dmso-d6: 3.97ppm, 3H, s, N-Me; 6.96ppm, 1H, s, уширен, C3H; 7.73ppm, 1H, s, уширен, C2H; 7.9-8.3ppm, 4H, 2d, J=5Hz, p-Ph; 8.71ppm, 1H, s, C6H.
Спектр ЯМР-19F, dmso-d6: 17ppm, 3F, s, -CF3; 25.5ppm, 3F, s, -CF3.
Масс-спектр: m/z 344 [M]+ (85).
1-Метил-4,7-вис-трифторметил-1Н-пирроло[3,2-c]пиридин (11).
Получали аналогично (10). Выделено 42мг (11) с выходом 87%. Медленно кристаллизующееся масло, Rf=0.9, элюент хлороформ/этилацетат в соотношении 9/1.
Спектр ЯМР-1Н, dmso-d6: 4.02ppm, 3H, s, N-Me; 6.9ppm, 1H, s, C3H; 7.9ppm, 1H, d, J=3Hz, C2H; 8.75ppm, 1H, s, C6H.
Спектр ЯМР-19F, dmso-d6: -53.5ppm, 3F, s, -CF3; -64.5ppm, 3F, s, -CF3.
Масс-спектр: m/z 268 [M]+ (71).
Список литературы
- Y.E. Pavlova, D.V. Gusev, A.S. Peregudov, N.D. Chkanikov., Synthesys of fused 3-trifluoromethylpyridines. Mendeleev Commun., 2006., 16(3), 177-178.
- (a) R. J. Tedeschi, Acetylene, in Encyclopedia of Physical Science and Technology, 3rd Edition, ed. R. A. Meyers, Acad. Press, Inc.: San Diego, 2001, vol. 1, p. 55-89; (b) Z. Wang Comprehensive Organic Name Reactions and Reagents. Part 3. Wiley, London, 2009, p. 2793-2796; (c) Name Reactions in Heterocyclic Chemistry. II, ed.: J. J. Li, Wiley-VCH, Hoboken, New Jersey, 2011, p. 72-82; (d) Б. A. Трофимов, A. И. Михалева, E. Ю. Шмидт, Л. Н. Собенина, Химия пиррола, Новые страницы, Наука, Новосибирск, 2012, 382 с; (е) Трофимов Б.А., Михалева А.И., Шмидт Е.Ю. и др., Пат. РФ N 2297410, Бюлл. Изобр. 2007. N 11.
- A.L. Sigan, D. V. Gusev, N.D. Chkanikov, E.Yu. Shmidt, A.V. Ivanov, A.I. Mihaleva., Hydroxyalkylation of 4,5,6,7-tetrahydroindole with polyfluorocarbonyl compounds as a route to 2-substituted indoles. Tetrahedron Letters, 52, (2011), 5025–5028.
- А.Н. Кост, М.А. Юровская, Ф.А. Трофимов, ХГС, 291, (1973). [Chem. Heterocyclic. Comp., 9, 267, (1973)].
- Р.С. Алексеев, А.В. Куркин, М.А. Юровская. γ-карболины и их гидрированные производные. 1. Ароматические γ-карболины: методы синтеза, химические и биологические свойства (обзор). ХГС, 2009, N8, с.1123-1166.
- Р.С. Алексеев, А.В. Куркин, М.А. Юровская. γ-карболины и их гидрированные производные. 2. Гидрированные производные γ-карболинов: методы синтеза (обзор). ХГС, 2010, N7, с.963-1018.
- (а) А.В. Бутин, А.С. Пилипенко, А.А. Милич, А.В. Финько, ХГС, 774, (2009). [Chem. Heterocycl. Comp., 45, 613, (2009)]; (b) Protective Groups in Organic Synthesis, Third Edition. Theodora W. Greene, Peter G.M. Wuts Copyright. 1999, John Wiley & Sons, Inc.
- Shumaila, Abdullah M. A.; Kusurkar, Radhika S.; Puranik, Vedavati G.; Tetrahedron; vol. 67; nb. 5; (2011); p. 936- 942.
- Yokoyama, Naota; Arai, Takayoshi; Chemical Communications (Cambridge, United Kingdom); nb. 22; (2009); p. 3285 – 3287.
- Вацуро К.В., Мищенко Г.Л., Именные реакции в органической химии., Москва, «Химия», 528 стр, 1976г.
- Р. С. Алексеев, А. В. Куркин, М. А. Юровская, Аза-γ-карболины и их бензаннелированные производные: методы синтеза, химические и биологические свойства., ХГС, 2011, N2, с.1765-1801.
- Яхонтов Л.Н., "Химия гетероциклических соединений", 1982, N 9, с. 1155-67.
- Evers Rainer; Klenz Oliver; Miethchen Ralf; Michalik Manfred., Organofluorine compounds and fluorinating agents Part 16: Monoalkylations and cycloadditions with trans-3,3,3-trifluoro-1 nitropropene Journal of Fluorine Chemistry, 1997 , vol. 81, # 2 p. 205 – 210.
Статья рекомендована к публикации членом редколлегии Д.х.н. Н.Д. Чканиковым
Fluorine Notes, 2014, 93, 3-4


