Поступило в редакцию: октябрь 2013
УДК 547.539.546.541.49
Fluorine Notes, 2014, 92, 1-2
РЕАКЦИИ АЛКИЛИРОВАНИЯ, АРИЛИРОВАНИЯ, АЛКЕНИЛИРОВАНИЯ И АЛКИНИЛИРОВАНИЯ ПОЛИФТОРАРЕНОВ И -ГЕТАРЕНОВ, ПРОТЕКАЮЩИЕ ПО АРОМАТИЧЕСКОМУ КОЛЬЦУ
Т.Д.Петрова, В.Е.Платонов
Федеральное государственное бюджетное учреждение науки Новосибирский институт органической химии им.
Н.Н.Ворожцова Сибирского отделения Российской академии наук, 630090 Новосибирск, проспект Акад. Лаврентьева,
9.
e-mail: petrova@nioch.nsc.ru
Аннотация. В обзоре рассмотрены реакции алкилирования, арилирования, алкенилирования и алкинилирования полифтораренов и – гетаренов, протекающие по ароматическому кольцу и приводящие к образованию связей СAr- C. Обзор содержит сведения о традиционных методах проведения таких превращений, которые включают, например, реакции полифтораренов с электрофильными, нуклеофильными, радикальными реагентами. Наряду с этими методами, рассматриваются реакции с применением металлокомплексного катализа. Применение металлокомплексного катализа расширяет область используемых субстратов. Этот метод включает реакции кросс-сочетания металло- и элементоорганических соединений и арилгалогенидов, катализируемые комплексами переходных металлов с различными органическими лигандами. Представлены реакции Кумады, Негиши, Сузуки, Стилла, Хийямы, Хека и Сононогаширы. В ряду полифтораренов и – гетаренов превращения связи CAr-Hal включает также и связь CAr-F.
Ключевые слова: полифторарены и – гетарены, алкилирование, арилирование, алкенилирование, алкинилирование, металлокомплексный катализ, палладиевые катализаторы, реакции кросс-сочетания.
I Введение
Образование новой С-С связи является одним из важнейших химических превращений органических соединений, поскольку позволяет осуществить синтез широкого круга веществ различного строения, которые по своим свойствам могут представлять интерес как сами по себе, так и в качестве исходных или промежуточных соединений в синтезе практически важных веществ. В ряду аренов для реакций с участием атома углерода ароматического кольца одним из путей образования связей СAr-С являются реакции алкилирования, арилирования, алкенилирования и алкинилирования. Эти превращения известны давно, и многие годы для их реализации использовались реакции с электрофильными, нуклеофильными и радикальными реагентами, внутримолекулярные перегруппировки и циклизации, трансформации уже имеющегося заместителя, приводящие к образованию алкил-, арил-, алкенил- или алкинильной группировок, а также реакции сочетания, катализируемые металлами (см., например, [1-3]. Однако, в последнее время наряду с этими традиционными методами стали развиваться и активно использоваться методы с применением металлокомплексного катализа с целью расширения границ используемых субстратов, оптимизации условий проведения процессов и выхода конечных продуктов, а также в тех случаях, когда обычные методы не давали желаемых результатов. К таким методам относятся реакции кросс-сочетания металло- и элементоорганических соединений и арилгалогенидов, катализируемые комплексами переходных металлов с различными органическими лигандами, что открывает широкие синтетические возможности и в настоящее время представляется одним из перспективных путей образования связи С-С. Реакция магнийорганических соединений носит название реакции Кумада, цинкорганических соединений– реакция Негиши, борорганических соединений – реакция Сузуки, оловоорганических соединений – реакция Стилла,кремнийорганических соединений – реакция Хияма, катализируемое палладием введение алкенильной группы носит название реакции Хека, а алкинильной – реакции Соногаширы. Рассмотрение закономерностей и механизмов этих реакций можно найти в прекрасных обзорах и монографиях, например [4-8].
В ряду полифтораренов и -гетаренов указанные выше тенденции развития методов образования связи СAr-С проявились в полной мере с тем лишь отличием, что для этих соединений трансформация связи СAr-Hal включает также и связь СAr-F.
Данные по реакциям образования связей СAr-С в полифтораренах, включая 1995 г, рассмотрены в обстоятельном обзоре Дж. Брука за 1997г [9]. В настоящей статье проанализированы литературные данные по реакциям алкилирования, арилирования, алкенилирования и алкинилирования полифтораренов и -гетаренов за последние ~ 10 лет. К рассматриваемым полифтораренам и –гетаренам отнесены соединения, содержащие два и более атома фтора в одном ароматическом кольце, а превращения включают трансформации связей СAr-Н и СAr-Hal, где Hal=I,Br,Сl,F. В статью включены также работы, касающиеся превращений других заместителей в полифторарене или –гетарене, но только непосредственно связанных с СAr, и только тех превращений, которые приводят к образованию алкильной или непредельной группировки. Реакции галоидалкилирования, в том числе перфторалкилирования, которым посвящены отдельные обзоры, в данную статью не входят и рассматриваются только в тех случаях, когда они представляют собой превращения интермедиатов на пути к конечному продукту.
II Превращения по связи СAr-Н
- Прямое введение алкил-, арил- и непредельных группировок
1.1. Реакции в присутствии кислотных катализаторовv
1.2. Термические и фотохимические превращения
1.3. Реакции в присутствии основания
1.4. Металлокомплексный катализ
1.4.1. Использование палладиевых катализаторов
1.4.2. Использование катализаторов на базе родия и никеля
2. Реакции с предварительной активацией связи СAr-Н металлированием
2.1. Образование и реакции литиевых производных
2.2. Образование и превращения медьорганических производных
2.3. Образование и превращения борорганических производных
2.3.1. Дифторфенилборные кислоты
2.3.2. Трифторфенилборные кислоты
2.3.3. Пентафторфенилборная кислота и полифторфенилбораты2.4. Образование и реакции полифторированных цинк- и оловоорганических соединений
1. Прямое введение алкил-, арил- и непредельных группировок
1.1 Реакции в присутствии кислотных катализаторов
К примерам меж- и внутримолекулярных реакций алкилирования полифтораренов типа реакций Фриделя-Крафтса относятся получение трис-(4-бром-2,3,5,6-тетрафторфенил)метана при взаимодействии 1-бром-2,3,5,6-тетрафторбензола с CHCl3 и AlCl3 (выход 22.4%) [10], хлорметилирование 1,3,5- трифторбензола действием на него ClCH2OCH3 в присутствии AlCl3 при кипячении в CS2 (80%) [11], а также внутримолекулярная циклизация под действием AlCl3 3-хлор-N-(3´,4´-дифторфенил)пропионамида до 6,7-дифтор-3,4-дигидро-1Н-хинолин-2-она [12,13]. Метил-3-(2´,6´-дифторфенил)-2-нитропропионат в большом количестве бензола, как растворителя, с избытком CF3SO3H легко подвергается фенилированию в положение 4´ с образованием бифенильного производного [14]. Трет.-бутилирование 3,5-дифторфенола в положение 4 протекает с выходом 54% при нагревании субстрата (55°С) с трет.-бутилметиловым эфиром в присутствии тетрахлорида циркония [15]. Модификация условий [16] не дала положительного результата (выход 31%). Перфторфенилирование перфторбензоциклоалкенов (перфторированные бензоциклобутен, индан, тетралин) и ряда их производных наблюдается при взаимодействии этих соединений с пентафторбензолом и п-перфторалкилтетрафторбензолами в среде SbF5 (схема 1) [17-23].
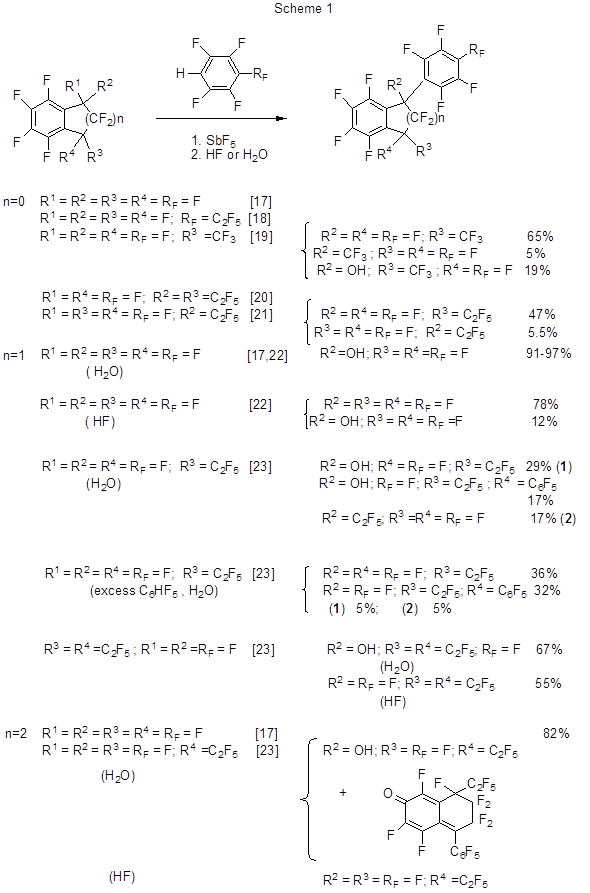
Реакция протекает за счёт образования из перфторбензоциклоалкенов под действием SbF5 катионоидных интермедиатов, которые взаимодействуют с полифторбензолами. Реакционная способность перфторбензоциклоалкенов при этом понижается по мере увеличения алициклического фрагмента [17]. Поскольку образующиеся полифторфенильные производные перфторбензоциклоалкенов в среде SbF5 существуют в виде солей соответствующих катионов выделение продукта из реакционной смеси осуществляется обработкой HF. При обработке водой получаются гидроксипроизводные за счёт взаимодействия последней с полифторированными катионами. Следует отметить, что в реакции перфтор-1,1-диэтилбензоциклобутена и пентафторбензола в присутствии эквивалентного количества SbF5 образующийся перфтор-1,1-диэтил-2-фенилбензоциклобутен выделить не удаётся, поскольку он быстро претерпевает дальнейшие превращения под действием SbF5 и Н2О, так что конечными продуктами оказываются главным образом триеноны (схема 2) [21].

Внутримолекулярные реакции Фриделя-Крафтса, сопровождающиеся образованием гетероциклического кольца, использованы для получения различных полифторированных хинолинонов, исходя из полифторированных анилидов со свободным орто-положением к амидной группировке. Так, нагревание 3,4-дифторанилида β-хлорпропионовой кислоты с AlCl3 при температуре 110°С [12] или 180°С [13] приводит к 6,7-дифтор-3,4-дигидро-1Н-хинолин-2-ону с выходом соответственно 78% и 37% (схема 3).
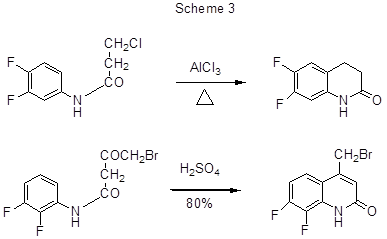
На схеме 3 приведён и другой пример электрофильной циклизации анилида [24].
Внутримолекулярное алкилирование, включающее циклизацию, имеет место при взаимодействии полифторированных анилидов коричной кислоты (ди-, три- и тетрафторпроизводных) с сильными кислотами, в частности, CF3 SO3H, или с AlCl3. В результате получаются полифторированные хинолиноны с достаточно высокими выходами (схема 4) [25-27].
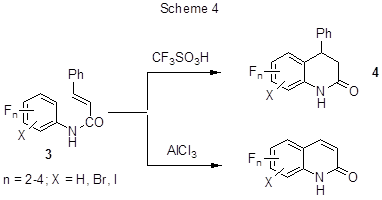
При этом реакции с AlCl3, протекающие в более жёстких условиях, сопровождаются
процессом дегидрофенилирования, который в аналогичных условиях наблюдается и в случае нефторированных
аналогов.
Интересно, что реакции циклизации идут, несмотря на дезактивирующее влияние нескольких атомов фтора. Предполагается [25], что превращение протекает через промежуточный суперэлектрофильный дикатион (5), образующийся в результате О,С-дипротонирования (схема 5). Лёгкость циклизации определяется влиянием заместителей как на равновесную концентрацию дикатиона 5, так и на энергию переходного состояния циклизации, которое в некоторой степени структурно подобно интермедиату (6).
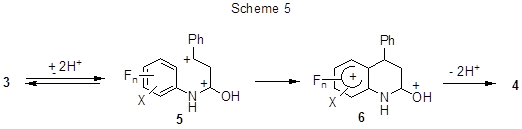
Однореакторный синтез полифторированных хинолинов по реакции Скраупа реализован при взаимодействии полифторанилинов, полифторгалогеноанилинов и полифторацетанилидов с глицерином (схема 6).
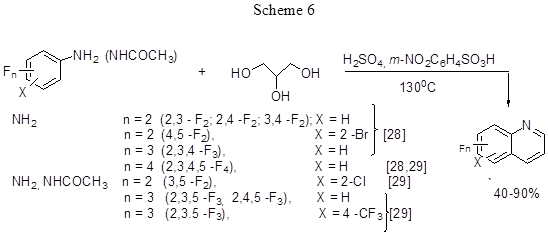
Реакция Фишера 2,5-дифторфенилгидразина с дизамещённым амидом 8-амино-1,4-диоксо-спиро[4,5]-декан-8-карбоновой кислоты ведёт к производным 5,8-дифтор-2,3,4,9-тетрагидро-1Н-карбазола (схема 7) [30].
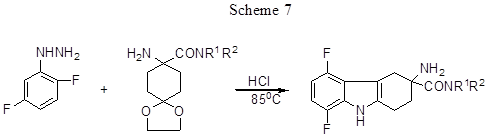
Ароматическая амино-кляйзеновская перегруппировка в ряду N-(1-метил-2-бутен-1-ил)производных фторированных анилинов в присутствии кислых катализаторов приводит к фторированным орто-алкиланилинам (схема 8) [31,32].
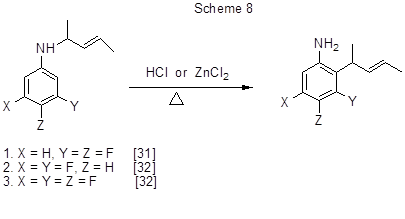
Реакция N-алкил-3,4-дифторанилина [31] (случай 1, схема 8) изучена более детально. В результате было установлено, что в качестве катализаторов могут быть использованы также эфират трёхфтористого бора и трифторуксусная кислота и что со всеми катализаторами помимо изомера, указанного на схеме 8, образуется и другой орто-изомер с алкильной группой в положении 6 ароматического кольца, а также продукт алкилирования в положение 6 исходного N-алкиланилина. Соотношение этих трёх продуктов меняется в зависимости от катализатора, но во всех случаях основным является соединение с алкильной группой в положении 2. Предполагается, что перегруппировка идёт не по согласованному [3,3] сигматропному механизму, а по межмолекулярному механизму, включающему электрофильный каталитический разрыв связи N-С1 и электрофильную атаку пент-3-ен-2-ильным катионом орто-положения ароматического кольца с последующим депротонированием σ-комплексов. Для 2,4,5-трифтор-N-(1-метил-2-бутен-1-ил)анилина перегруппировка не идёт [32], что связывают с дезактивацией положения 6 атомом фтора в положении 5.
Амино-кляйзеновская перегруппировка протекает при взаимодействии избытка 3,4-дифторанилина с 1-хлор-2-циклопентеном. Первоначально образующееся N-циклопентенильное производное под действием хлоргидрата исходного анилина превращается в смесь орто-циклопентенильных производных дифторанилина с преобладанием 6-изомера (схема 9) [33].

Реакция этого анилина и 1,4-бензохинона с предварительным диазотированием дифторанилина приводит с выходом 90% к продукту дифторфенилирования 1,4-бензохинона в положение 2 [34].
1.2. Термические и фотохимические превращения
Термическая внутримолекулярная реакция Дильса-Альдера эфира 2,4-дифторкоричной кислоты, содержащего фрагмент пропаргиловой кислоты, даёт возможность синтезировать дифтордигидронафтофураноновые производные (схема 10) [35].
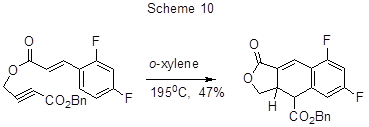
Термическая перегруппировка Кляйзена аллиловых эфиров полифторированных фенолов
в присутствии N,N-диметиланилина позволяет получать полифторированные орто-аллилфенолы (схема
11) [36-38].
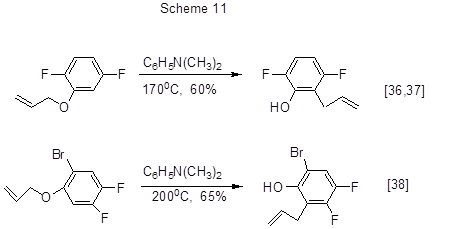
Удобным одностадийным методом алкилирования дезактивированных к реакциям Фриделя-Крафтса
полифторированных бензольных карбонильных производных или бензонитрилов являются фотоинициируемые
цепные радикальные реакции с галогенидами алкилртути [39]. Так, окислительное трет-бутилирование
дифторзамещённых бензольных производных успешно осуществляется путём фотолиза их смеси с t-BuHgCl
в присутствии основания – 1,4-диазобицикло[2,2,2]октана (ДАБСО) как акцептора протонов (схема 12).
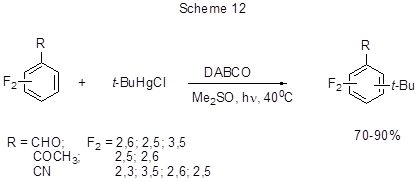
Механизм протекания процесса, по-видимому, включает участие трет-бутильного радикала в процессе бимолекулярного ароматического гомолитического замещения с образованием радикального аддукта и далее - анион-радикала, образующегося в результате отщепления основанием (ДАБСО) протона из радикального аддукта. И радикальный аддукт, и анион-радикал стабилизируются заместителем R. Цепная реакция инициируется переносом электрона от анион-радикала к t-BuHgCl, регенирирующим трет-бутильный радикал (схема 13).
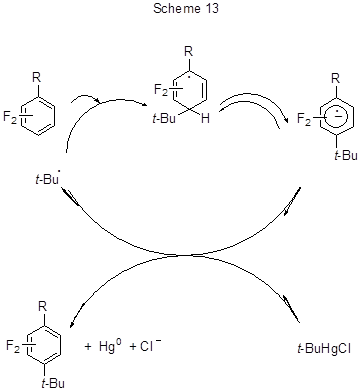
Реакции трет-бутилирования протекают преимущественно в пара-положение к R, и когда оно занято, реакция не идёт. В соответствии с приведённой схемой 13 региоселективность должна определяться резонансной стабилизацией радикального аддукта, образующегося за счёт орто- или пара-атаки трет-бутильным радикалом. Однако, орто-атака менее выгодна из-за образования в качестве интермедиата стерически напряжённого радикала с объёмной трет-бутильной группой в орто-положении. Тем не менее, в случае 2,4-дифторбензонитрила, хотя пара-положение и занято, реакция идёт, и с выходом 46% получается орто-трет-бутильное производное дифторбензонитрила. Таким образом, орто-замещение по отношению к нитрильной группе активируется, возможно, за счёт вклада в процесс линейной резонансной структуры (схема 14), в которой стерическое напряжение уменьшается. Дифторацетофеноны обычно менее реакционноспособны, чем аналогичные нитрилы или альдегиды, возможно из-за стерически напряжённой ацильной группы в промежуточном радикальном аддукте.
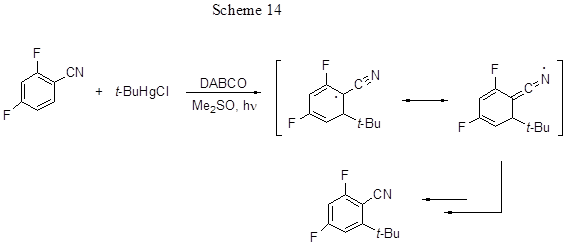
Метод окислительного сочетания 2-нафтола с 5,6,7,8-тетрафтор-2-нафтолом в присутствии медного катализатора позволяет получить с умеренным выходом тетрафторбинафтол (схема 15) [40].
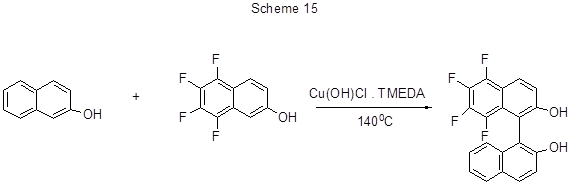
С другими катализаторами реакция не идёт. Для этого превращения очень важны температура
реакции (не ниже 125°С) и количество катализатора (не более 10%). В других условиях образуется бинафтол,
не содержащий атомов фтора.
1.3. Реакции в присутствии оснований
Трет-бутилацетатная группировка была введена в 4-е положение 2,6-дифторнитробензола реакцией 2,6-дифторнитробензола с трет-бутилхлорацетатом в присутствии трет-бутилата калия (схема 16) [41).

Викариозное нуклеофильное замещение в 2,6-дифторнитробензоле с трет-бутилэтилмалонатом и NaH в DMF с последующей обработкой CF3COOH приводят к этиловому эфиру 2,3-дифтор-4-нитрофенилуксусной кислоты [42].
1.4. Металлокомплексный катализ
Указанные в предыдущих разделах типы реакций, в особенности различные модификации реакций Фриделя-Крафтса, равно как и другие превращения, ведущие к образованию новой С-С связи, успешно использовались в органической химии и позволили осуществить синтез алкил-, арил-, алкенил- и алкинил-производных соединений различных классов, в том числе аренов и гетаренов, однако, ряд ограничений, характерных для такого типа реакций, инициировал работы по созданию новых подходов к получению производных указанных типов. Одним из наиболее перспективных и успешно развиваемых в последнее десятилетие подходов к образованию С-С связи, открывающем широкие синтетические возможности, является использование катализа комплексными катализаторами на базе переходных металлов. Применение катализаторов для трансформирования инертных С-Н связей особенно важно для тех случаев, когда известные методы неэффективны или неэкономичны. Превращения с участием металлокомплексного катализатора включают как прямое замещение атома водорода С-Н связи на желаемую группу, так и предварительное замещение его на металл с образованием металлоорганического соединения и последующее взаимодействие последнего с реагентом, содержащим нужную группировку. Обычно это реакции кросс-сочетания арена или металларена с соответствующими галогенопроизводными.
В общем виде такие превращения можно представить следующим образом:
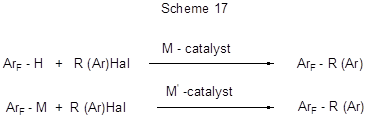
Далее будет рассмотрено использование этих подходов в ряду полифторированных аренов и гетаренов.
1.4.1. Использование палладиевых катализаторов.
Такой путь перспективен для прямого арилирования электронодефицитных полифтораренов, поскольку обычные реакции с электрофильными реагентами для таких соединений затруднены. Авторы работ [43,44] предлагают новые условия прямого арилирования пента-, тетра-, три- и дифторбензолов, 2,3,5,6-тетрафторанизола, 2,3,5,6-тетрафтортолуола, а также 4Н-тетрафторпиридина реакцией кросс-сочетания с широким набором арил- и гетарилгалогенидов (Hal = I,Br,Cl) в присутствии основания (K2CO3, 1.1 equiv.) и палладиевого катализатора Pd(OAc)2 (1-5 mol %) и P(t-Bu)2Me – HBF4 (2-10 mol %) или образующегося in situ из Pd(OAc)2 и 2-дициклогексилфосфино-2´,6´-диметоксибифенила (S-Phоs) (схема 18).
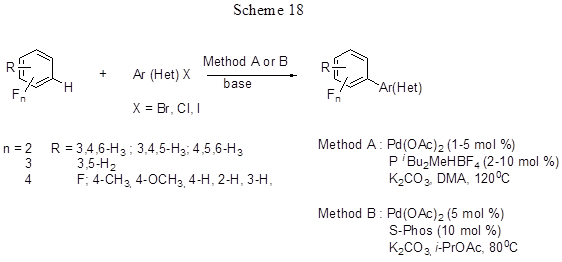
Реакция с пентафторбензолом идёт при 80°С в изопропилацетате, с другими полифтораренами –в DMA при 120°С. Ранее успешное арилирование пентафторбензола осуществлялось путём кросс-сочетания пентафторфенильных производных бора с арилиодидами (подробнее о реакциях такого типа см. в разделе 2.3.). В данном же случае нет необходимости получать заведомое пентафторфенилпроизводное бора, что упрощает схему процесса и, безусловно, относится к достоинствам метода. При взаимодействии полифтораренов с хлорбромбензолом в реакции участвует связь С-Br. С дибромаренами наблюдается двойное арилирование. Хорошо идёт реакция с бромпроизводными арена, имеющими различные заместители как в одном, так и в обоих орто-положениях. Хлорарены, как активированные пара-метильной группой, так и дезактивированные, активно вступают в реакцию. В случае иодпроизводных лучшие результаты получаются при добавлении небольшого количества Ag2CO3 [44], которое служит для удаления образующихся солей иодидов, ингибирующих процесс. Три- и дифторбензолы реагируют с меньшим выходом, чем тетра- и пентафторбензолы (выходы с пентафторбензолом составляют 65-98%, с дифторбензолами – 29-42%), т.е. в реакции наблюдается инверсия реакционной способности полифторированных аренов по сравнению с обычным электрофильным ароматическим замещением, т.к. более кислые С-Н связи арена реагируют предпочтительнее. Если фторарен содержит несколько атомов водорода, может идти ди- и триарилирование, и получается смесь продуктов. Для 1,3-дифторбензола реакция идёт по наиболее кислому протону в положении 2 (выход продукта 85%). Исследование конкурентных реакций показало, что реакционная способность С-Н связи фторарена параллельна её кислотности, и при наличии двух С-Н связей реакция идёт по более кислой, которая находится в орто-положении к фтору. Возможные пути протекания реакций показаны на схеме 19 на примере арилирования пентафторбензола арилбромидом. В соответствии с расчётными данными [43] реакция не включает образование металлоциклических интермедиатов. Ключевая стадия – расщепление связи С-Н, включающая согласованное металлирование фторарена и перенос Н либо к карбонатному лиганду катализатора (С2 путь), либо к лиганду Br (C1 путь). Чувствительность металлирования напрямую зависит от кислотности разрываемой С-Н связи. Наиболее низкий энергетический барьер имеет путь С2 и он лучше совпадает с экспериментальными данными, поэтому он предполагается как наиболее вероятный, хотя путь С1 также не может быть полностью отвергнут и, возможно, реализуется в ряде случаев.
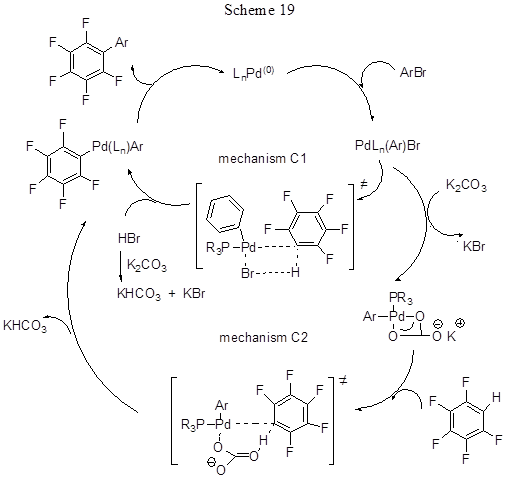
Рассмотренные реакции масштабируемы и при использовании примерно равных эквимольных количеств двух арильных компонент ведут к полифторированным бифенильным продуктам, которые могут быть важны для медицинской химии, электрон-транспортных устройств, органических светопроводящих диодов и элементов в дизайне жидких кристаллов [43]. Сходные условия арилирования пентафторбензола с образованием пентафторбифенильных производных использованы в реакции с бромфенильным фрагментом ряда С-2´-дезоксирибонуклеотидов (выход 64% [45]) и 3-(трифторметилацетокси)-4-метоксибромбензолом (выход 53% [46] ).
Интересный вариант реакции прямого арилирования пентафторбензола действием п-бромтолуола с использованием Pd-катализатора был предложен в работе [47]. Как и в выше рассмотренных работах, реакция проводилась в присутствии основания (в данном случае К3РО4) при 120°С (растворитель DMF) и с катализатором Pd(OAc)2, не содержащем в качестве лигандов объёмных электронодонорных фосфинов, что предпочтительно с точки зрения экономики и производства, но как сокатализатор добавлялась триметилуксусная кислота (pivalic acid). Карбоксилат-анион способствует понижению энергии расщепления С-Н связи и может служить переносчиком протона от субстрата к неорганическому основанию, а вместе – катализатор и сокатализатор- осуществляют процесс металлирования-депротонирования, например, как на схеме 20, иллюстрирующей механизм процесса арилирования ксантинов действием п-бромтолуола в присутствии Pd(OAc)2 и С(СН3)3СООН. Выход продукта арилирования пентафторбензола таким путём составил 80%.

Окислительным сочетанием 1,4-дифторбензола с 1,2-нафтохиноном в присутствии Pd(OAc)2 и СН3СООН по методике [48], описанной для 1.4-дихлорбензола, был получен 4-(2,5-дифторфенил)-1,2-нафтохинон (схема 21) [49].

Активация С-Н связи в 3,4-дифторацетанилиде палладиевым комплексом имеет место при окислительной реакции Мизороки-Хека этого анилида с бутилакрилатом [50]. Основным направлением реакции является алкенилирование в положение 6 (выход продукта 60%). При наличии в положении 2 триметилсилильной группы образуется 2-алкенильное производное (85%) (схема 22).
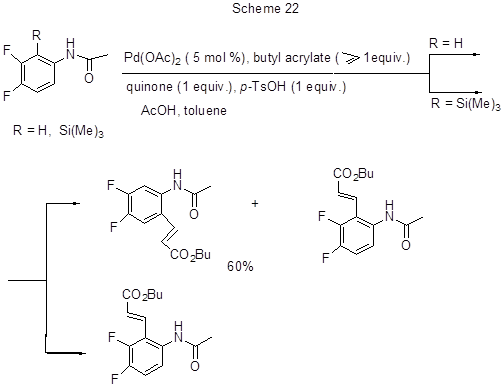
Реакция идёт через образование палладиевых комплексов циклического характера путём замещения палладием орто-атомов водорода или кремния соответственно. Эти комплексы были выделены и охарактеризованы спектральными и рентгеноструктурными методами. В указанных превращениях наличие ацетамидогруппы обязательно, т.к. она облегчает присоединение электрофильного палладия к кольцу и направляет образование новой С-С связи в орто-положение. Действительно, со свободными аминами реакция не идёт.
Взаимодействие дигалогенанилинов с галогензамещёнными пиридинами в присутствии палладиевого катализатора и основания в однореакторном процессе приводит к α-карболинам. Реакция идёт в две стадии через палладий-катализируемое ариламинирование с последующим внутримолекулярным арилированием. Таким путём был получен 5,8-дифтор-α-карболин (схема 23) [51].
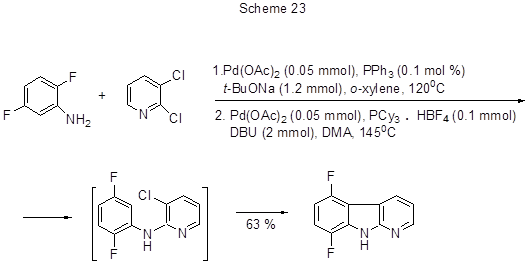
Оптимальными являются условия, когда на первом этапе в качестве лиганда для палладиевого катализатора используется трифенилфосфин, а далее добавляется ещё комплекс трициклогексилфосфина с НВF4 , 1,8-диазобицикло[5.4.0]ундец-7-ен (DBU) и повышается температура реакции.
Катализируемое палладием исключительно 5-exo-dig гидроарилирование с внутримолекулярной циклизацией до производных 9-бензилиден-9Н-флуорена наблюдается в случае орто-алкинилбиарилов (схема 24) [52].
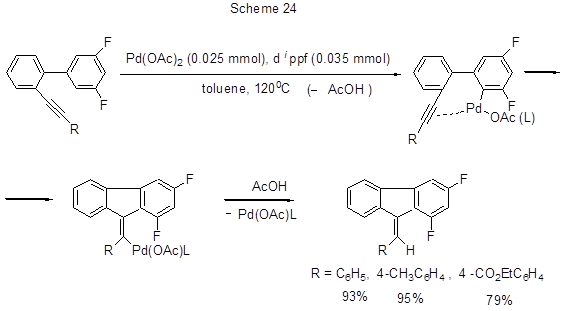
Реакция протекает с активацией связи СAr-Н путём орто-паллаидирования,
внедрения палладия в тройную связь с образованием винилпалладиевого интермедиата, который в результате
протодепаллаидирования превращается в конечное соединение с регенерацией катализатора.
Ещё одним удобным вариантом палладий-катализируемого метода прямого арилирования электронодефицитных полифтораренов является реакция их кросс-сочетания с арилборными кислотами в присутствии окислителя (схема 25) [53].
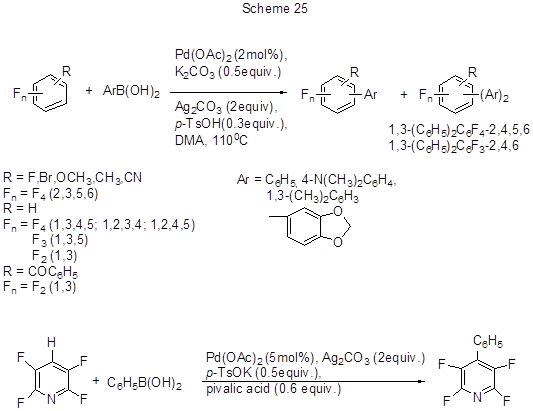
В реакциях с пентафторбензолом, помимо указанных арилборных кислот, использовались также кислоты с Ar = 4-RC6H4 (R = Cl, CH3, OCH3, CHO, N(CH3)2). Успех реакции обусловлен одновременным применением в качестве добавок к Pd(OAc)2 и окислителю слабого основания, которое способствует расщеплению кислой С-Н связи, и слабой кислоты, которая уменьшает скорость трансметаллирования арилборных кислот, приводящего к их нежелательному гомосочетанию. На примере реакции пентафторбензола (0.2 ммол) с фенилборной кислотой было установлено, что оптимальные условия образования пентафторбифенила (выход 90%) – это использование 2 мол % Pd(OAc)2, 0.5 экв. К2СО3, 0.3 экв. пара-толуиловой кислоты и 1.2 экв. фенилборной кислоты. Эффективный окислитель – Ag2CO3 (2 экв.), температура реакции 110°С , растворитель DМА. Донорные заместители в молекуле арилборной кислоты способствуют протеканию реакции образования биарила, но с орто-метилзамещённой фенилборной кислотой реакция сочетания не идёт. Меньшая реакционная способность арилборных кислот с электроноакцепторными заместителями может быть компенсирована заменой основания (например, использованием KF вместо K2CO3 ) и увеличением количества этого основания. Реакционная способность полифтораренов прямо зависит от кислотности С-Н связи, и ею определяется выбор основания. Для превращения более кислых С-Н связей 2,3,5,6-тетрафторпиридина и 2,3,5,6-тетрафторбензонитрила использовали 0.5 экв. К-соли п -толуиловой кислоты как основания и 0.6 экв. триметилуксусной кислоты при соотношении полифторарена и арилборной кислоты 3:1. В случаe полифтораренов с менее кислыми С-Н связями (изомерные тетрафторбензолы, бромтетрафторбензол, 1,3,5-трифторбензол, 1,3-дифторбензол и 2,4-дифторбензофенон) использовали комбинацию 0.5 экв. К3РО4 и 0.6 экв. триметилуксусной кислоты. Хотя в молекуле некоторых полифтораренов потенциально имеется несколько положений для арилирования, в основном реализуется моноарилирование по наиболее кислой С-Н связи, например, между двумя атомами фтора. Так, 1,3-дифторбензол арилируется в положение 2. В случае 1,3,5-трифторбензола получается смесь моно- и диарильного производных в соотношении 4.5 : 1, с 1,3,4,5-тетрафторбензолом - смесь моно- и дипроизводных в соотношении 4.6 : 1. Арилирование же 2,4-дифторбензофенона идёт исключительно в мета-положение к бензоильной группе и пара- положение к фтору.
Авторы предлагают следующую схему превращения, включающую первоначальное образование арилпалладиевого интермедиата путём транс- металлирования В→Рd с последующим паллаидированием фторарена путём согласованного металлирования-депротонирования и восстановительного элиминирования Pd(0) (схема 26).
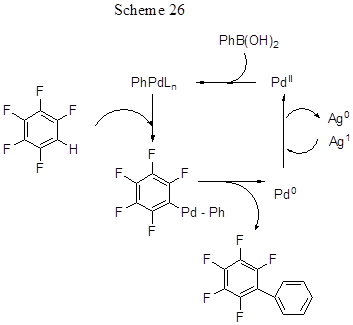
1.4.2. Использование катализаторов на базе родия и никеля.
Производные полифторарилборной кислоты также могут быть использованы в качестве арилирующих агентов, например, для хинонов. Так, при взаимодействии трифтор-(3,5-дифторфенил)бората с 1.5-мольным избытком нафтохинона и каталитическим количеством родиевого катализатора получается 2-дифторфенильное производное нафтохинона, однако выход соединения не высок (22%, схема 27) [54]. В качестве побочного продукта получается 1,4-гидроксинафталин.

Реакция 3,4-дифторфенилборной кислоты с 1-(1-бензил-2,5-дигидро-1Н-пиррол-3-ил)этаноном в присутствии родиевого катализатора идёт путём присоединения фторированного арильного фрагмента по двойной связи 2,5-дигидропиррольного кольца (схема 28) [55].
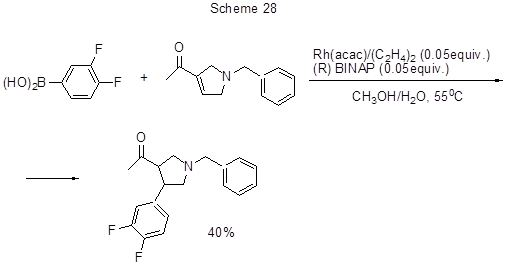
Аминокляйзеновская перегруппировка N-пропаргиланилинов с акцепторными заместителями в кольце, включая атомы фтора, протекает при нагревании в гексафторизопропиловом спирте и с родиевым катализатором, давая 2- или 2,3-замещённые индолы [56]. Этот подход был использован для синтеза 2,3-диметил-4,6-дифториндола (схема 29). Предполагается, что реакция протекает с промежуточным образованием орто-аллениланилинового производного в качестве интермедиата за счёт собственно аминокляйзеновской перегруппировки, которое в результате внутримолекулярной циклизации переходит в производное индола.

Реальным катализатором в процессе является алкоксильный аддукт RhH(CO)(Ph3P)3 и растворителя, который был выделен и охарактеризован (схема 30).

Эффективное алкенилирование и алкинилирование полифтораренов за счёт преимущественной активации связи СAr –Н по сравнению с СAr- F происходит при их взаимодействии соответственно с 1,3-диенами, виниларенами и алкинами в присутствии комплексного никелиевого катализатора [57]. Преимущество метода состоит в том, что нет необходимости предварительного депротонирования кислого водорода полифторарена действием стехиометрического количества органометаллического основания с последующей реакцией с электрофилом. Для активации связи СAr-Н в этом случае достаточно применения каталитических количеств (5-10 мол%) никилиевого катализатора. Интересно, что хотя обычно для комплексов Ni (0) более предпочтительно окислительное внедрение по связи С-F, нежели С-Н, в данном случае в реакции участвует связь С-Н (схема 31).
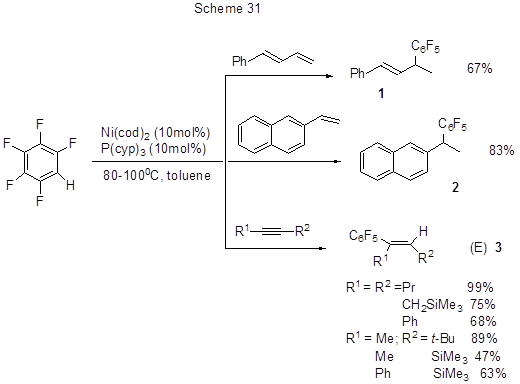
Стерически напряжённые несимметричные алкины присоединяются стереоселективно, давая
цис-аддукты с объёмными заместителями исключительно в транс-положении к полифторированному кольцу.
В случае 4-октина изучены также реакции с 2,3,5,6-тетра- и 3,5-дифторпиридином, 1,2,3,4-, 1,2,3,5-
и 1,2,4,5-тетрафторбензолом, 1,2,3-трифторбензолом и 1,2- и 1,3-дифторбензолами, 2,3,5,6-тетрафторанизолом
и метиловым эфиром 3,5-дифторбензойной кислоты. При наличии в полифторарене нескольких атомов водорода
реакция идёт и по второй СAr -Н связи с образованием диалкенилированных полифтораренов,
причём соотношение моно- и дипродуктов зависит от соотношения реагентов – избыток 4-октина повышает
дизамещение, а при избытке полифторарена преобладает моно-алкенилирование. Увеличение числа атомов
фтора способствует протеканию реакции. Наиболее активированы С-Н связи в орто-положении
к фтору.
Реакция с 1-фенил-1,3-бутадиеном идёт по терминальной двойной связи (схема 31). При взаимодействии с 2-винилнафталином (схема 31) получается 1,1-диарилэтан. Механизм реакций на примере пентафторбензола представлен на схеме 32.
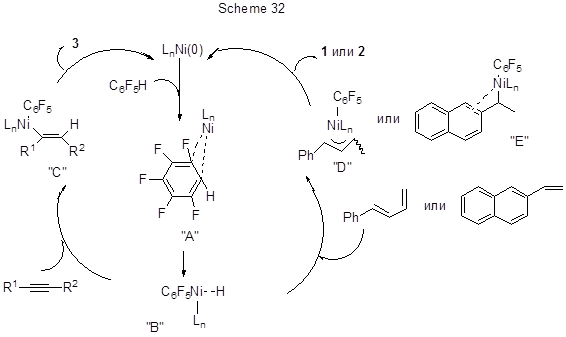
Реакция начинается с образования η2-аренового комплекса “А” с последующим окислительным присоединением Ni к С-Н связи арена и образованием “В”. Внедрению алкина в связь Ni-H предшествует координация, позволяющая избегнуть стерического отталкивания между наиболее объёмной группой R2 и C6F5–кольцом у Ni-центра. Восстановительное элиминирование даёт алкенилированный полифторарен (3) и регенерирует Ni(0) соединение. В случае алкенов образуются соответственно π-аллильный комплекс “Д” или бензилникелевый комплекс “Е”, которые при восстановительном элиминировании дают продукты (1), (2) и регенерируют Ni(0).
2. Реакции с предварительной активацией связи СAr-Н металлированием.
2.1. Образование и реакции литиевых производных.
Хорошо известным и широко используемым методом введения в ароматическую молекулу алкильных и арильных групп является замещение ароматического атома водорода на литий и последующее взаимодействие образующегося литий-органического соединения с электрофилом. Наличие атомов фтора в молекуле полифторарена делает соседние протоны более кислыми, что облегчает их взаимодействие с литиевыми основаниями. Литированию подвергается наиболее кислый протон в орто-положении к атому фтора. В случае двух атомов фтора в мета-положении литированию подвергается связь СAr-Н между этими атомами фтора. Реакция литирования низкотемпературная и, как правило, проводится в присутствии N,N-диизопропиламина или N,N,N´,N´-тетраметил-1,2-диаминоэтана (TMEDA). Таким путём в реакциях с иодистым метилом осуществлено метилирование 2,4,5-трифторбензойной кислоты до 2,4,5-трифтор-3,6-диметилбензойной кислоты и 3-хлор-2,4,5-трифторбензойной кислоты до 3-хлор-2,4,5-трифтор-6-метилбензойной кислоты [58,59], трет.-бутил-(2,3-дифторбензилокси)-диметилсилана до 4-метилпроизводного [60], 2,4-дифторбромбензола до 3-метил-2,4-дифторбромбензола [61], 3,5-дифторбромбензола до 4-метил-3,5-дифторбромбензола [62], 1-циклопропил-6,7-дифтор-2-метокси-1Н-хиназолин-2,4-диона до 5-метильного производного [63]. Действием иодистого метила на 1,3-дифтор-5-(3´, 5´- диметоксибензилокси)бензол с выходом 90% получают 2-метильное производное, которое аналогичным путём метилируется до 2,4-ди- и 2,4,6-триметильного производного (схема 33) [64].
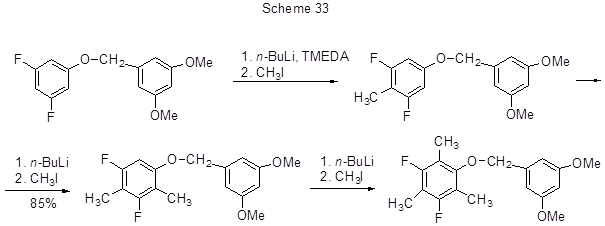
В данном случае положение литирования определяется не только орто-ориентирующим эффектом атома фтора, но и комплексообразованием кислорода с литиевым реагентом, однако первый эффект преобладает и определяет место вступления первой метильной группы (положение 2). Этому способствует и мета- расположение атомов фтора и кислорода. Для соединений, где расположение другое, селективно литируется бензильный протон, хотя и с низким выходом.
Другие примеры моно- или диметилирования полифторарильных фрагментов с предварительным литированием и действием метилиодида описаны в работах [65, 66], с диметилсульфатом – в работе [67]. 2,3-Дифторалкилбензолы (Alk = n-Bu, i-Bu, Et, n-Pr, циклогексилметил) были получены из о-дифторбензола в реакции с н-бутиллитием и соответствующим алкилиодидом в присутствии TMEDA [68]. Литирование 3,4-дифторбромбензола в положение 2 и последующая реакция с трифлатом циклопентенового ряда с хорошим выходом приводит к образованию продукта с бензильной С-С связью (схема 34) [69].
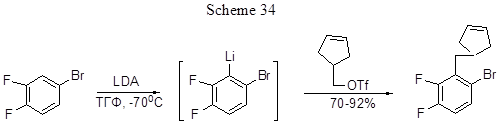
Некаталитическое анионное сочетание ариллитиевых производных с 1,2-дибромбензолом даёт прямой выход к орто, орто-галогенозамещённым биарилам, которые могут иметь практическое значение, поскольку биарильный фрагмент входит во многие биологически активные соединения, используются в фармацевтических и жидкокристаллических структурах. Такая реакция реализована взаимодействием м-дифторбензола [70] и ряда его производных [71] с 1,2-дибромбензолом (схема 35).
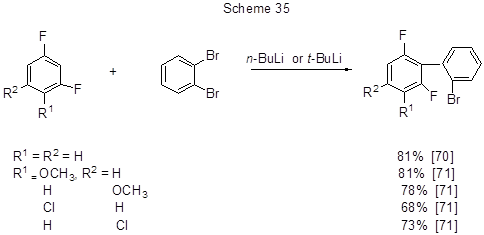
Механизм превращения предполагает образование ариллитиевых интермедиатов обменом Hal→М и Н→М и участие в процессе высоко реакционноспособного дегидробензола, который получается превращением нестабильного литиевого производного из о-дибромбензола. Далее идёт нуклеофильное присоединение к дегидробензолу достаточно стабильного полифторированного литиевого производного, и эта стадия является скоростьопределяющей. Вслед за этим происходит перенос брома от исходного о-дибромбензола, и получается конечный орто-полигалогенoамещённый биарил (схема 36). Достоинством метода является отсутствие сложных катализаторов для сочетания.

2-Бром-2',6'- дифторбифенил был получен аналогичным путём из 1,3-дифторбензола и о-бромхлорбензола (выход 66%) [72].
Образование промежуточного дегидробензола предполагается и из продукта литиирования 2,6-дифтортриметилсилилбензола Этот дегидробензол реагирует с литиевыми предшественниками, давая изомерные трифторбифенилы (схема 37) [73].
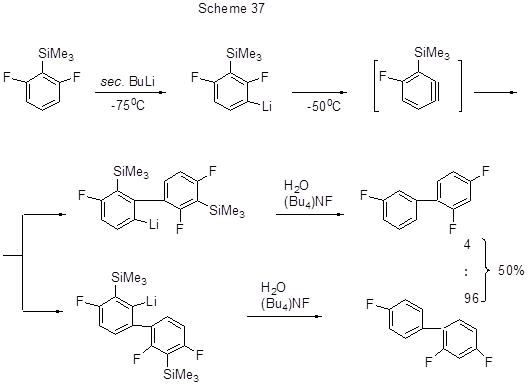
Реализована также реакция циклоалкилирования о-дифторбензола действием 4-(транс-4-пропилциклогексил)-циклогексана с одновременным дегидрированием, так что конечным продуктом является 2,3-дифтор-1-[транс-4-(транс-4-пропилциклогексил)циклогексенил]бензол (выход 48%, схема 38) [74].
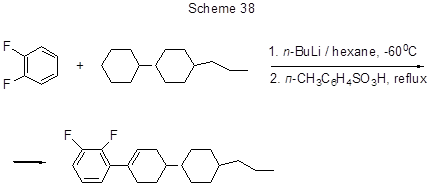
Литийорганические соединения, образующиеся из полифтораренов, могут присоединяться по карбонильной группе альдегидов или кетонов с образованием продуктов оксиалкилирования полифтораренов. Некоторые примеры таких превращений приведены на схеме 39.
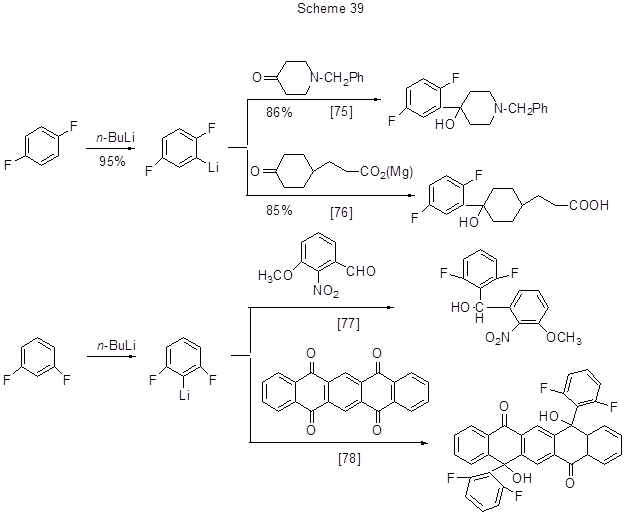
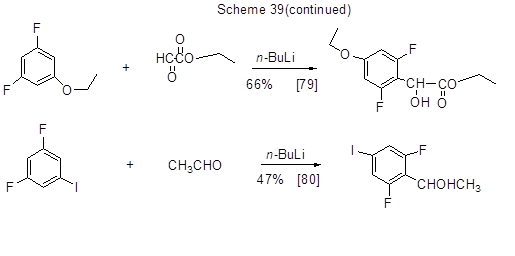
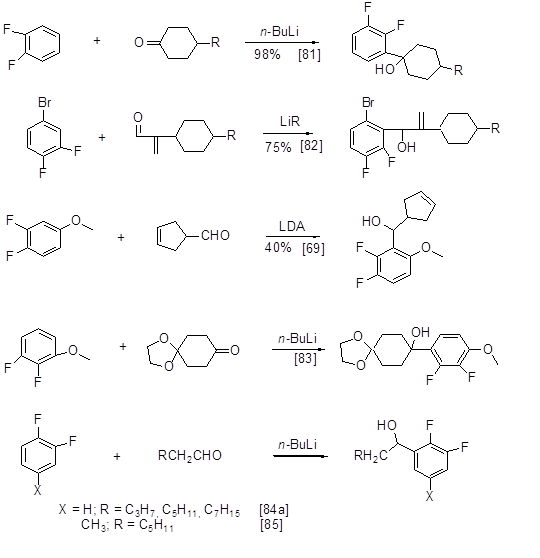
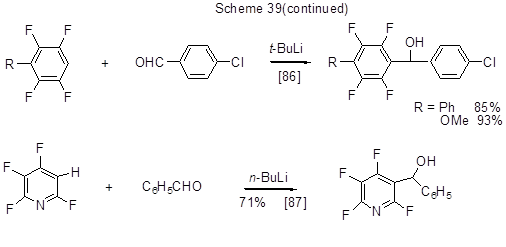
Помимо приведённых выше превращений полифторированных литийаренов такие литийарены часто используются в металлорганическом синтезе как исходные для получения в ходе процесса переметаллирования других металлорганических производных, в частности, соединений меди, бора и др.
2.2. Образование и превращения полифторированных медьорганических соединений.
Общий метод катализируемого медью арилирования, бензилирования и алкенилирования Сsp2–H связи, разработанный для широкого круга соединений с рКа ниже 35, успешно применён в ряду полифтораренов. Он включает взаимодействие аренов, содержащих два и более атомов фтора в кольце, с соответствующими галогенопроизводными в присутствии оснований, катализируемое солями меди [88-91]. В качестве галогенопроизводных пригодны как иодиды, так и бромиды, в некоторых случаях и хлориды, например, гетероциклические, которые могут содержать как донорные, так и акцепторые группировки. В случае менее реакционноспособных соединений (например, мезитиленового производного) лучше использовать иодид. Наилучшие результаты получаются при комбинации CuI, как катализатора, с фенантролиновым лигандом. Наиболее приемлемое основание – К3РО4, но для дифторзамещённых субстратов лучшие результаты получаются с алкоксидами лития. Растворитель – DMF или DMF\ксилол. Этот метод – удобная и более дешёвая альтернатива катализу другими металлами, например, палладиевыми, рутениевыми или родиевыми катализаторами (схема 40).

Наиболее эффективно реакция идёт по наиболее кислой С-Н связи между двумя атомами фтора. В случае 1,2,3,4-тетрафторбензола, где таких связей нет, реакция с п-иодтолуолом протекает только с 10%-ным выходом [89]. При наличии в молекуле галогенопроизводного двух одинаковых [89,90] или разных галогенов [91] в реакции участвуют оба эти галогена, и таким образом в молекулу вводятся две арильные группировки Ar. Вероятный механизм реакции представлен ниже (схема 41).
Scheme 41
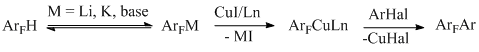
Предполагается, что под действием основания идёт депротонирование кислой связи СAr – Н и образование ArFM с последующим трансметаллированием до медьорганического соединения, которое сочетается с ArHal, давая конечный продукт. В подтверждение схемы в реакции с пентафторбензолом был получен, выделен и охарактеризован промежуточный комплекс C6 F5Cu –фенантролин, который реагирует с арилиодидом, давая биарил [89].
Оказалось, что возможна прямая каталитическая реакция сочетания полифторированных аренов и с соединениями, не содержащими атомов галогена. Так, взаимодействие полифторарена с терминальными алкинами в присутствии медного катализатора на воздухе позволяет получать с выходом от 25 до 76% полифторарилалкины (схема 42) [92].
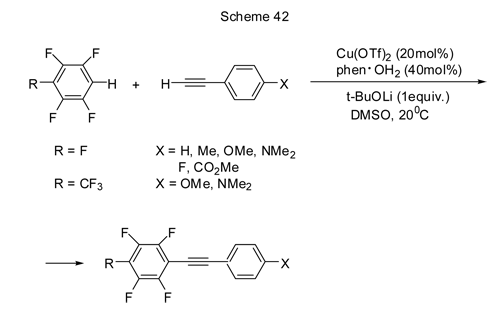
Наличие донорных заместителей Х способствует повышению выхода продукта сочетания. В случае R=CF3 даже с донорным Х выходы ниже, чем с R=F. Три- или дифторарены не вступают в эту реакцию, что может быть связано с недостаточной кислотностью С-Н связи в этих соединениях. Не реагируют таким образом и алкилалкины.
2.3. Образование и превращения полифторарилборорганических соединений.
Арилборорганические соединения, обычно арилборная кислота или её производные, получаемые из соответствующих ариллитиевых производных и соединений бора, вступают в реакции кросс-сочетания с арилгалогенидами, трифлатами или тозилатами, протекающие в присутствии основания и катализируемые комплексами переходных металлов, чаще всего палладия или никеля. При этом происходит отщепление галогена или OSO2R-группировки, и образуется новая С-С связь за счёт сочетания фрагментов арил-арил.

Это превращение, называемое реакцией Сузуки, открывает большие синтетические возможности и представляет один из наиболее перспективных методов получения би- и полиарилов, многие из которых обладают практически полезными свойствами или служат основой для получения соединений с такими свойствами [93]. В общем виде реакция может быть представлена следующим образом (схема 43) [5, p.25, 26]. Хлориды наименее активны в этой реакции вследствие более высокой энергии диссоциации связи С-Сl, что ведёт к пониженной способности к окислительному присоединению Pd(0) или Ni(0) к этой связи, а эта стадия является ключевой во всём процессе. Как и во всех превращениях с металлокомплексным катализатором, в реакции Сузуки важную роль играет выбор основания, а также лигандов для катализатора и количество используемого катализатора. Эта реакция со всеми её особенностями, достоинствами, недостатками и механизмом подробно рассмотрена как в ряде обзоров, например, [4, 94], так и монографий [5-8]. В данном разделе проанализировано использование её в ряду полифтораренов, причём в число рассмотренных полифторбораренов включены соединения, литиевые предшественники которых получены как обменом на литий протона связи СAr-Н, так и галогена связи СAr-Hal. Подробнее о превращениях связи СAr-Hal →CAr-Li см. в разделе III.
2.3.1. Дифторфенилборные кислоты.
2,4-Дифторфенилборная кислота (1) и её 3,4- (2), 3,5- (3) и 2,5- (4) дифторизомеры реагируют с 3-бромфенилметилацетатом в присутствии NaOH и трифенилфосфинового производного палладия как катализатора с замещением брома [95]. Аналогично протекает реакция (1) с 4-бромбензальдегидом с этим же катализатором и К2СО3 в качестве основания (схема 44) [96].
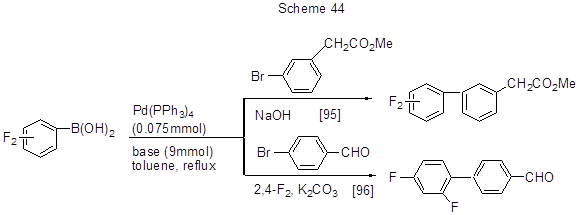
При взаимодействии 1 с метил-2-{[(трифторметил)сульфонил]окси}бензоатом был получен метил-2´, 4´-дифторбифенил-2-карбоксилат (схема 45) [97].
Реакция Сузуки кислоты 1 с 4-бромацетофеноном проводилась в присутствии К2СО3 и различных комплексных палладиевых катализаторов, давая продукт кросс-сочетания с высоким выходом [98, 99].
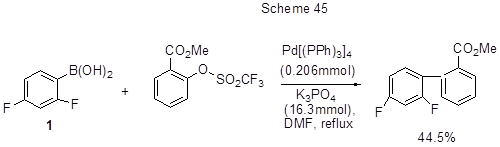
Оказалось, что если в качестве лиганда для PdCl2 взять электронодонорный, стерически объёмный дибензил-N-(диизопропил)фосфорамидит P(OBn)2N(i-Pr)2 , а Na2CO3 в качестве основания, то можно использовать только 0.01 мол % катализатора, проводить реакцию при комнатной температуре и получать продукт с выходом 92% [100].

Примеры реакций соединения 1 с гетероциклическими галогенопроизводными, содержащими атом галогена непосредственно в гетероцикле или в арильной компоненте гетероциклического соединения, приведены на схеме 46 (звёздочкой обозначен замещаемый атом галогена). Палладиевыми катализаторами в большинстве случаев являются Pd(PPh3)4 или PdCl2(PPh3)2, а основаниями - K2CO3, Na2CO3, Cs2CO3.
Другие примеры таких реакций соединения 1 с различными гетероциклическими галогенидами см. [107-115]. Процесс с использованием микроволнового облучения описан в работе [116]. Все эти реакции, как правило, представляют собой одну из стадий многоступенчатых процессов получения сложных веществ с потенциально полезными свойствами.
Реакции Сузуки с 3,5-дифторфенилборной кислотой 3 иллюстрирует схема 47.

3,5-Дифтордифенилметаны получаются с высоким выходом (70-97%) при взаимодействии
соединения 3 с бензилхлоридом или бромидом и катализатором Pd[(N-succ.)Br(PPh3)2]
[121]. Полифторированные алкилциклогексильные производные би- или терфенилов получаются кросс-сочетанием
по Сузуки кислоты 3 и 4- или 4´- алкилциклогексилпроизводных 1-иод-2-фторбензола
или 3-фтор-4-иодбифенила соответственно в присутствии Pd/C и K2CO3 (выходы
от 65 до 87%) [122, 123]. Пример замещения трифлатной группировки в производном циклогексена на 1,3-дифторфенильную
группу в реакции с 3 приведён в работе [124] (выход продукта 55%). Реакция 2-гидрокси-3,5-дифторфенилборной
кислоты с 2-замещённым 4-бром-1,3-тиазолом идёт с замещением брома, но выход 4-дифторарильного производного
1,3- тиазола не высокий (16%) [125].
Кислота 3 была использована также для получения биарилов и гетеробиарилов другим методом, который реализуется как однореакторное превращение смеси 3 с бензолом или тиофеном в присутствии Mn(III)-ацетата при микроволновом облучении (схема 48) [126].
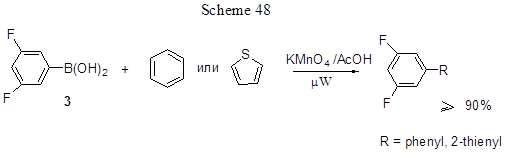
Это обычный радикальный путь получения биарилов, а Mn(OAc)3 . 2H2O – одноэлектронный окислитель, используемый для генерации радикалов, что ведёт к образованию С-С связи. Он может быть получен из KMnO4 + CH3COOH при микроволновом облучении. Новизна предложенного метода заключается в использовании микроволнового облучения вместо обычного нагрева, а его преимущества заключаются в достижении высокого выхода продуктов, короткого времени реакции и использовании малых количеств бензола или тиофена, которые берутся только как реагенты, но не растворители.
С целью получения соединений интересных по своим медико-биологическим свойствам осуществлен синтез, на начальных стадиях которого использована реакция Сузуки енолтрифлата замещённого пиразола с 2,3-дифторфенилборной кислотой (5) [113], аналогичная реакциям дифторфенилборных кислот 1, 2, 3 и 2,3,5-трифторфенилборной кислоты [113], протекающая с замещением OTf-группировки.
В плане получения веществ с интересными жидко-кристаллическими свойствами изучена реакция Сузуки галогеноаренов и кислоты 5, а также её 4-алкокси- и 4-алкилпроизводных, в т.ч. содержащих длинные алкильные цепи, приводящая к полифторированным бифенилам с конечными алкоксильными и алкильными группировками. Реакции кислоты 5 с бром- [84b] и иодаренами [81, 93] представлены на схеме 49, а её 4-алкоксипроизводных с длинными алкильными цепями – на схеме 50 [81, 84b, 127, 128].
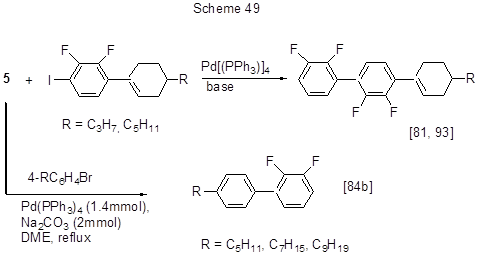
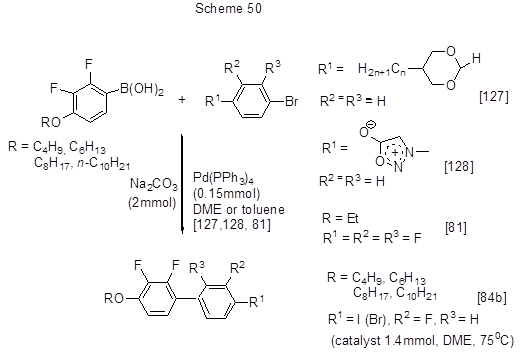
Вовлечение в реакцию 4-алкилпроизводных кислоты 5 привело к различным результатам. Так, реакция сочетания 2,3-дифтор-4-гептил-6-метилборной кислоты с 2-циано-4-оксиоктил-6-метилбромбензолом в присутствии Pd(PPh3)4 не шла, и только путём использования 3 mol % PdCl2, 6 mol % PPh3, CsF в качестве основания и тщательного высушивания всех реагентов удалось получить соответствующий бифенил с выходом 21% (схема 51) [85]. В модельном опыте с бромбензолом выход составлял 40%.

Кросс-сочетание 4-алкилпроизводных кислоты 5 с бромфенолом или его эфирами [68], а также с 1-иод-2-фтор-4-бромбензолом представлены на схеме 52.
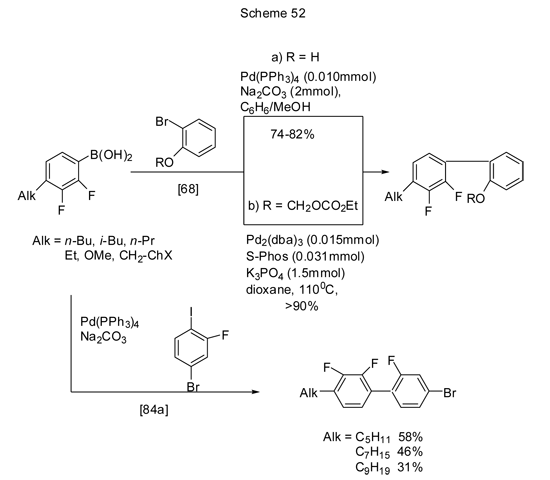
В плане создания терфенилов нужной конструкции с желаемым расположением заместителей, исходя из 4-алкильных или 4-арильных производных кислоты 5, синтезировано две серии терфенилов с терминальными алкильными цепями, содержащими два атома фтора в концевом или центральном кольце, при этом в первом случае в реакцию в качестве галогеноарильной компоненты вводится бромпроизводное биарилa (схема 53) [84а].

Наконец, на схеме 54 представлен пример использования пара-триметилсилильного производного эфира кислоты 5 в реакции Сузуки, приводящей при постепенном наращивании цепи к полифторированным пара-кватерфенилам [129]. Поскольку при проведении описанных в работе [129] реакций использовались комбинаторные методики, а также галогенопроизводные с различным количеством атомов фтора или вообще без фтора, в результате был получен широкий набор бифенилов, тер- и кватерфенилов, содержащих разное число атомов фтора. Использование более устойчивых эфиров борных кислот вместо свободных кислот позволяет уменьшить побочный процесс деборилирования. Следует отметить, что в изученных реакциях Сузуки, если имеется более чем два атома фтора в орто-положении к образующейся С-С связи, выход продуктов получается низкий или реакция не идёт вообще.
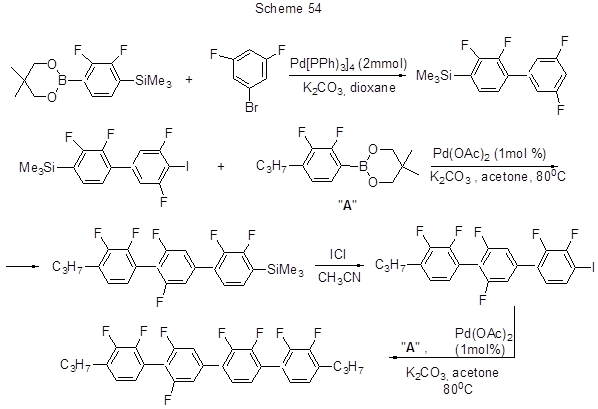
Об образовании бифенильных производных в реакции Сузуки борной кислоты 2 с 2-нитро-4-хлор-метилбензоатом или о-бромфенолом см. [130-132], с 3,5-замещённым бромбензолом [133]. Соответствующий бифенил получается с выходом 94% из кислоты 2 и 4-(4´-алкилциклогексил)бромбензола при использовании в качестве катализатора комплекса бис(имино)пиридин Pd (II) [134]. На схеме 55 приведены примеры реакции Сузуки кислоты 2 с гетероциклическими соединениями, содержащими атомы брома и трифлатную группировку, а также орто-N-пивалоиламидного производного кислоты 2 с трифлатом циклопентенового ряда.

Нуклеофильное присоединение кислоты 2 к бензальдегиду в присутствии никелевого катализатора приводит к оксиалкильному производному (схема 56) [139]. Механизм превращения пока не выяснен, но предполагается, что никелевый катализатор играет роль кислоты Льюиса.
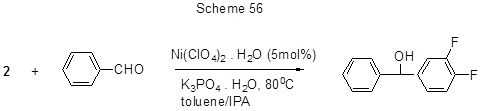
Реакция с гетероциклическим трифторметансульфонатом, аналогичная приведённой на
схеме 55, реализована и для фенилборной кислоты 4 [137]. Эта же кислота вступает
в реакцию Сузуки с хлорпроизводным замещённого 1,3-оксазола [117], п-бромтолуолом с применением
микроволнового облучения [140] и о-броманилином [141].
2,6-Дифторфенилборная кислота (6) [104], подобно другим изомерным дифторфенилборным кислотам, 4-замещённая кислота 6 [142, 143] и эфир 4-замещённой кислоты 6 [123] также вступают в реакцию Сузуки.
2.3.2. Трифторфенилборные кислоты
3,4,5-Трифторфенилборная кислота (7) реагирует по реакции Сузуки с 7-замещённым 3-иодхромоном в присутствии Pd/C и K2CO3 , давая с 80%-ным выходом соответствующий хромон с трифторфенильной группой в положении 3 [114,115]. Реакция с участием атома хлора реализуется в случае 7-хлор-4-окса-2-пропил-1,2,3,4,4а,9,10,10а-октагидрофенантрена (катализатор – PdCl2(PCy3)2 ) [144]. Бромпроизводные также легко вступают в реакции кросс-сочетания с кислотой 7 по Сузуки. Так, 4-(1-оксициклопентил-1)бромбензол и кислота 7 в присутствии Pd(PPh3)4 , (Bu)4NBr и NaHCO3 дают трифторбифенильное производное с выходом 90% [145]. 4-(4´-Алкилциклогексил)бромбензол и кислота 7 реагируют по Сузуки при использовании бис(имино)пиридинпалладий (II) комплекса как катализатора с образованием бифенильного производного , имеющего в одном ароматическом кольце 4-( 4´-алкилциклогексил)-группу и три атома фтора в другом, с выходом 94% [134]. В случае замещённого бифенила, имеющего в каждом кольце по атому брома [146], или гетероциклического дибромпроизводного, приведённого на схеме 57 [147], в реакции участвуют оба атома брома.
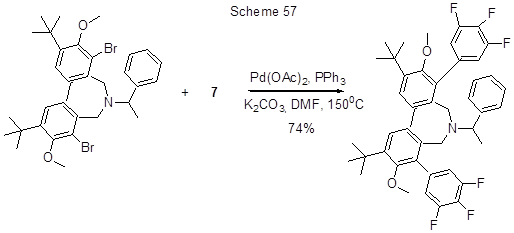
Наконец, реакция Сузуки кислоты 7 и 6-оксо-8-пропил-7,8,9,10-тетрагидро-6Н-бензо[c]-хромен-3-ил трифторметансульфоната с PdCl2(PPh3)2 и водным гидразин-гидратом идёт с участием OTf-группы [148].
2,3,4-Трифторфенилборная кислота использована в реакции кросс-сочетания с о-гидроксибромбензолом [132] с целью получения соединений с орто-гидроксибифенильной системой, перспективных в качестве непептидных ингибиторов протеазы (накопление атомов фтора способствует возрастанию ингибиторной активности). 5-Алкил- и 5-алкоксипроизводные 2,3,4-трифторфенилборной кислоты вводили в реакцию Сузуки для синтеза полифторированных терфенилов, содержащих 1,5-дизамещённые 2,3,4-трифторфенильные фрагменты, которые обладают свойствами жидких кристаллов (схема 58) [149].
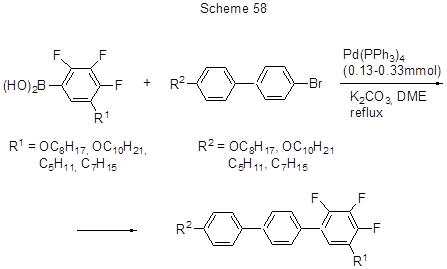
Реакции идут с высоким выходом.
О кросс-сочетании по Сузуки 2,3,5-трифторфенилборной кислоты и трифлата 3-оксоциклогекс-1-енила, протекающим с участием трифлатной группы, см. [150].
2.3.3. Пентафторфенилборная кислота и полифторфенилбораты.
Пентафторфенилборная кислота в обычных условиях кросс-сочетания по Сузуки не активна, но использование в качестве добавок к палладиевому катализатору комбинации Ag2O и CsF промотирует реакцию [151]. Наиболее эффективно (выходы более 90%) реакция осуществляется с ароматическими иодидами или бромидами, соответственно с С6Н5I и XC6H4Br, где X= o-, м-, п-CH3, м-, п-ОСН3, п-F, п-ОС2Н5, п- CF3, п-NO2, 2-нафтил. В случае хлорбензола выход пентафторбифенила составляет только 39%. Оптимальной каталитической системой для иодидов является Pd(PPh3)4 / CsF /Ag2O, а для бромидов (и хлорбензола) Pd2(dba)3 / P(t-Bu)3 / CsF / Ag2O.
В работах Фрона и Бардина [152] приведены примеры использования литиевых и калиевых солей полифторфенилборатов в качестве реагентов для получения бифенильных производных в катализируемых палладием реакциях кросс-сочетания с ароматическими иодпроизводными в присутствии стехиометрических количеств Ag2O. Общая схема превращений представлена ниже (схема 59).
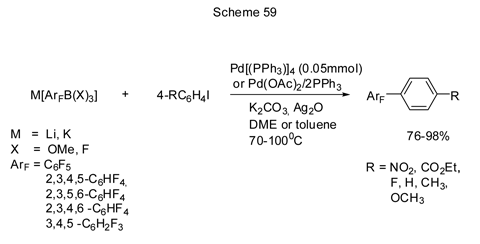
Литиевые соли, в принципе, более реакционноспособны, чем калиевые, но добавка Ag2O выравнивает реакционную способность обоих типов солей в этом превращении. Замещённые бромбензолы оказались менее эффективными, чем иодпроизводные [152 b]. С невысоким выходом получаются бифенильные производные и при взаимодействии K[C6F5BF3] с замещёнными бензолдиазонийтетрафторборатами [4R-C6H4N2][BF4]. Лучшим палладиевым катализатором при этом оказался Pd(PPh3)4 [152c].
Сочетание калиевой соли трифтор-(2,6-дифторфенил)бората с 2-замещённым 5-иодпиридином в присутствии PdCl2dppf даёт с выходом 17% продукт замещения иода на 2,6-дифторфенильную группу [153].
2.4. Образование и реакции полифторированных цинк- и оловоорганических соединений
Кросс-сочетанием по реакции Негиши цинкового комплекса, полученного из 1,2,3-трифторбензола, с 3-бромтиофеном синтезирован 3-(3,4,5-трифторфенил)тиофен. Катализатор – PdCl2(PPh3)2 или Pd(PPh3)4 [154]. В работе [155] для получения реагентов арил- или гетероарилцинка из активированных аренов и гетаренов с последующим использованием этих реагентов в реакции сочетания Негиши предложено новое, мягкое и эффективное основание – 2,2,6,6-тетраметилпиперидинцинк хлорид – литий хлорид (TMPZnCl .LiCl), которое в свою очередь получается из 2,2,6,6-тетраметилпиперидина с BuLi и ZnCl2 (схема 60).
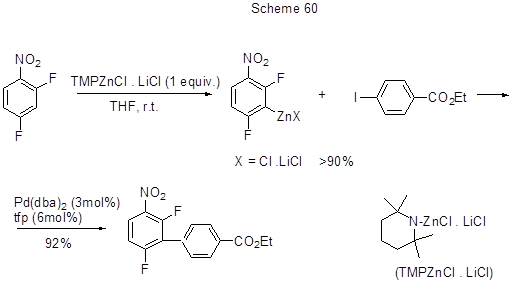
Последовательное взаимодействие 1,3-дифторбензола или 1,3-дифтор-5-метоксибензола с n- BuLi и ZnCl2 и далее с (3-амино-2-бромпиридин-4-ил)-(2,4-дифторфенил)метаноном в присутствии Pd(PPh3)4 позволяет получать дифторарены с пиридиновым фрагментом (схема 61) [112]. Следует отметить, что конечный продукт с R=H может быть получен и по реакции Сузуки при использовании 2,6-дифторфенилборной кислоты 6 [112].
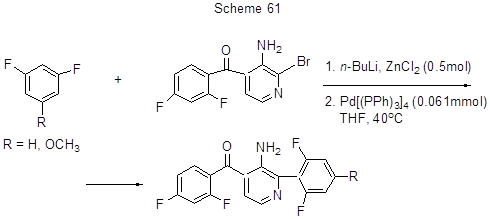
Предполагается, что реакция идёт с промежуточным образованием цинковых производных дифтораренов, которые при взаимодействии с пиридиновым бромпроизводным по реакции Негиши переходят в наблюдаемые продукты. Сходным образом идёт 3,4,5-трифторфенилирование 4´-(3-н-бутилбицикло[1,1,1]-пент-1-ил)-3,5-дифторбифенила в положение 4 при взаимодействии его с 3,4,5-трифторбромбензолом, n- BuLi, ZnCl2 и Pd(PPh3)4 [156].
Модификация синтеза полифторированных бифенилов по реакции Негиши состоит в предварительном получении полифторалюминийарильных производных прямым алюминированием с помощью алюминиевого реагента, которые далее путём транс-металлирования с ZnCl2 превращаются в цинковые производные. Последние вступают в палладий-катализируемое кросс-сочетание по Негиши с арилиодидами, приводя к бифенильным производным (схема 62) [157].

Одной из стадий получения потенциальных антидепрессантов ряда биарилкарбамоилиндолинов является реакция Стилла, заключающаяся во взаимодействии (2,6- или 3,5-дифторфенил)трибутилолова с 5-бромникотинатом (схема 63) [158].
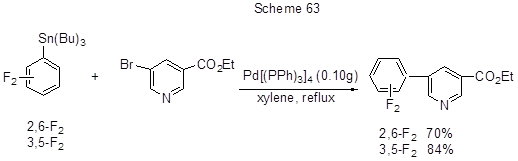
продолжение в следующем номере
Список сокращений
Список литературы
[2] Mоррисон Р., Бойд Р. Органическая химия., Мир, Москва, (1974).
[3] Бартон Д., Оллис В.Д. Общая органическая химия, Т.1., Химия, Москва, (1981).
[4] Hassan J., Sevignon M., Gozzi C., Schulz E., Lemair M., Chem. Revs., (2002), 102, 1359-1470.
[5] Ed. Ackermann L. Modern Arylation Methods., John Wiley, Weinheim, (2009).
[6] Смит В.А., Дильман А.Д. Основы современного органического синтеза., БИНОМ, Лаборатория знаний, Москва, (2009).
[7] Eds. Meijere A., Diederich F. Metal-Catalyzed Cross-Coupling Reactions., John Wiley, Weinheim, (2004).
[8] Schlosser M. Organometallics in Synthesis, 2nd Ed., John Wiley, Chichester, (2002).
[9] Brooke G.M., J. Fluor. Chem., (1997), 86, 1-76.
[10] Filler R., Fiebig Jr. A.E., Mandal B.K., J. Fluor. Chem., (2000), 102, 185-188.
[11] Koda T., Yasntake M., Shinmyozu T., Org. Lett., (2001), 3, 1419.
[12] Tabart M., Picaut G., Desconclois J-F., Dutka-Maben S., Huet Y., Berthaud N., Bioorg. Med. Chem. Lett., (2001), 11, 919-922.
[13] Lundbeck H., Pat. WO 106534 (2009) (C.A., 2009, 151, 337044).
[14] Nakamura S., Sugimoto H., Ohwada T., J. Am. Chem. Soc., (2007), 129, 1724- 1732.
[15] Pat. US 211741 (2006) (C.A., 2006, 145, 377029).
[16] Pat. US 270682 (2006) (C.A., 2007, 146, 27833).
[17] Карпов В.М., Меженкова Т.В., Платонов В.Е., Синяков В.Р., Щёголева Л.Н., Ж. орган. хим., (2002), 38, 1210-1217.
[18] Karpov V.M., Meshenkova T.V., Platonov V.E., Sinyakov V.R., J. Fluor.Chem., (2002), 117, 73-81. Karpov V.M., Meshenkova T.V., Platonov V.E., Sinyakov V.R.,Shchegoleva L.N., Russ. J. Org. Chem.,(2002), 38, 1158-1165 (English translation).
[19] Синяков В.Р., Меженкова Т.В., Карпов В.М., Платонов В.Е., Рыбалова Т.В., Гатилов Ю.В., Ж. орган. хим., (2003), 39, 886-891. Sinyakov V.R., Meshenkova T.V., Karpov V.M., Platonov V.E., Rybalova T.V., Gatilov Y.V., Russ. J. Org. Chem.,(2003),39, 837-842 (English translation).
[20] Zonov Ya.V., Karpov V.M., Platonov V.E., J. Fluor.Chem., (2007), 128, 1058- 1064.
[21] Синяков В.Р., Меженкова Т.В., Карпов В.М., Платонов В.Е., Ж. орган. хим., (2007), 43, 1678-1686. Sinyakov V.R., Meshenkova T.V., Karpov V.M., Platonov V.E., Russ. J. Org. Chem.,(2007),43, 1677-1685 (English translation).
[22] Karpov V.M., Mezhenkova T.V., Platonov V.E., Sinyakov V.R., J. Fluor. Chem., (2001), 107, 53-57.
[23] Синяков В.Р., Меженкова Т.В., Карпов В.М., Платонов В.Е., Рыбалова Т.В., Гатилов Ю.В., Ж. орган. хим., (2006), 42, 85-93. Sinyakov V.R., Meshenkova T.V., Karpov V.M., Platonov V.E., Rybalova T.V., Gatilov Y.V., Russ. J. Org. Chem.,(2006),42, 77-86 (English translation).
[24] a) Bonnefous C., Payne J.E., Roppe J., Zhuang H., Chen X., Severance D., Noble S.A., Smith N.D., Symons K.T., Nguyen P.M., Shiau A.K., Hassig C.A., Sablad M., Rosenkrants N., Zhang Y., Tadimeti S., Wang L., Rix P., Walsh J.P., Yazdani N., J. Med. Chem., (2009), 52, 3047-3062; b) Pat. WO 29592 (2009) (C.A., 2009, 150, 283051); c) Pat. WO 29617 (2009) (C.A., 2009, 150, 282846); d) Pat. WO 29625 (2009) (C.A., 2009, 150, 283049).
[25] Safina L.Yu., Selivanova G.A., Shteingarts V.D., Koltunov K.Yu., Tetrahedron Letters., (2009), 50, 5245-5247.
[26] Inglis S.R., Stojkoski C., Branson K.M., Cawthray J.F., Fritz D., Wiadrowski E., Pyke S.M., Booker G.W., J. Med. Chem., (2004), 47, 5405-5417.
[27] Breault G., Eyemann C.J., Geng B., Momingstar M., Reck F., Pat. WO 134378 (2006) [C.A. (2007), 146, 81779].
[28] Kato T., Saeki K., Kawazoe Y., Hakura A., Mutation Res., (1999), 439, 149- 158. [29] Laev S.S., Gurskaya L.Yu., Selivanova G.A., Beregovaya I.V., Shchegoleva L.N., Vasil’eva N.V., Shakirov M.M., Shteingarts V.D., Eur. J. Org. Chem., (2007), 306-316.
[30] Shuttleworth S.J., Lizarzabaru M.E., Chai A., Coward P., Bioorg. Med. Chem. Letters., (2004), 14, 3037-3042.
[31]Мустафин А.Г.,Гимадиева А.Р.,Халилов И.Н.,Спирихин Л.В.,Фатыхов А.А.,Нурушев Р.А.,Абрахманов И.Б.,Толстиков Г.А.,Изв.АН.Сер.хим., (1998), 188-190. Mustafin A.G., Gimadieva A.R., Khalilov I.N., Spirikhin L.N., Fatykhov A.A., Nurushev R.A., Abdrakhmanov I.B., Tolstikov G.A., Bull. Russ. Acad. Sci. Div. Chem.Sci., (1998), 47, 188-190 (English translation).
[32]Гатауллин Р.Р.,Миннигулов Ф.Ф.,Кудашев А.Р.,Нурушев Р.А.,Абрахманов И.Б.,Ж.прикл.хим., (2002), 75, 95-97. Gataullin R.R., F. F. Minnigulov F.F., Kudashev A.R., Nurushev R.A., I. B. Abdrakhmanov I.B., Russ. J. Applied Chem., (2002), 75, 95-97 (English translation).
[33]Гатауллин Р.Р.,Кажанова Т.В.,Давыдова В.А.,Исмагилова А.Ф.,Зарудный Ф.С.,Фатыхов А.А.,Спирихин Л.В.,Абрахманов И.Б.,Хим.-фарм.ж. (1999), 33, 29-32. Gataullin R.R., Kazhanova T.V., DavydovaV.A., IsmagilovaA.F., Zarudii F.S., Fatykhov A.A., SpirikhinL.V., I.B. AbdrakhmanovI.B., Russ. J. Chem. Pharm., (1999), 33, , 255-258.
[34] Song Yu., Zhu A., Lv J., Gong G., Xie J., Zhou J., Ye Y., Zhong X., Spectrochim. Acta Part A: Molecular and Biomol. Spectroscopy, (2009), 73, 96-100.
[35] Chackalamannil S., Doller D., Clasty M., Xia Y., Fagen K., Lin Y., Hsung-An Tsai, McPhail A.T., Tetrahedron Letters, (2000), 41, 4043-4047.
[36] Yoshimura H., Kikuchi K., Hibi Sh., Tagami K., Satoh T., Yamauchi T., Ishibahi A., Tai K., Hida T., Tokuhara N., Nagai M., J. Med. Chem., (2000), 43, 2929-2937.
[37] Pat. US 6110959 (2000) (C.A., 1997, 127, 278135); Pat. US 6355669 (2002) (C.A., 1999, 130, 306588); Pat. US 6258811 (2001) (C.A., 1998, 128, 270532).
[38] Pat. US 7045545 (2006) (C.A., 2000, 133, 135218).
[39] Kim B.H., Jeon I., Lee D.B., Park H.J., Jun Y.M., Baik W., J. Fluor. Chem., (2001), 109, 145-149. 106
[40] Yekta S., Krasnova L.B., Mariampillai B., Picard C.J., Chen G., Pandiaraju S., Yudin A.K., J. Fluor. Chem., (2004), 125, 517-525; Pat. WO 51899 (2005) (C.A., 2005, 143, 27355).
[41] Pat. WO 60160 (2009) (C.A., 2009, 150, 313452).
[42] Pat. US 78249 (2003) (C.A., 2001, 134, 86149).
[43] Lafrance M., Rowley Ch.N., Woo T.K., Fagnou K., J. Am. Chem. Soc., (2006), 128, 8754-8756.
[44] Lafrance M., Shore D., Fagnou K., Org. Letters, (2006), 8, 5097-5100.
[45] Youbert N., Urban M., Pohl R., Hocek M., Synthesis, (2008), 1918-1932.
[46] Litvinas N.D., Brodsky B.H., DuBois J., Angew. Chem., Int. Ed.Engl., (2009), 48, 4513-4516.
[47] Zhao D., Wang W., Lian S., Yang F., Lan J., You J., Chemistry – A Europ. J., (2009), 15, 1337-1340.
[48] Itahara T., J. Org. Chem., (1985), 50, 5546-5550.
[49] Ahn J.H., Cho S.Y., Ha J-D., Chu S.Y., Jung S.H., Jung Y.S., Baek J.J., Choi I.K., Shin E.Y., Kang S.L., Kim S.S., Cheon H.G., Yang S.D., Choi J.K., Bioorg. Med. Chem. Letters, (2002), 12, 1941-1946.
[50] Rauf W., Thompson A.L., Brown J.M., Chem. Commun., (2009), 3874-3876.
[51] Laha J.K., Petron Ph., Cuny G.D., J. Org. Chem., (2009), 74, 3152-3155.
[52] Chernyak N., Gevorgyan V., J. Am. Chem. Soc., (2008), 130, 5636-5637; Advanced Synth. and Catalysis, (2009), 351, 1101-1114.
[53] Wei Y., Kan J., Wang M., Li W., Hong M., Org. Letters, (2009), 11, 3346- 3349.
[54] Demchuk O.M., Pietrusiewicz K.M., Synlett, (2009), 18, 1149-1153.
[55] Pat. US 42896 (2009) (C.A., 2009, 150, 237419).
[56] Saito A., Oda S., Fukaya H., Hanzawa Yu., J.Org. Chem., (2009), 74, 1517- 1524.
[57] Nakao Y., Kashihara N., Kanyiva K.S., Hiyama T., J. Am. Chem. Soc., (2008), 130, 16170-16171.
[58] Pat. US 6211375 (2001) (C.A., 1997, 127, 331407).
[59] Pat. US 6136823 (2000) (C.A., 1998, 129, 41087).
[60] Pat. US 6235764 (2001) (C.A., 2000, 132, 22961).
[61] Pat. US 6348624 (2002) (C.A., 1999, 131, 73434); Pat. US 6329391 (2001) (C.A., 2002, 136, 20031).
[62] Pat. EP 1223169 (2002) (C.A., 1996, 125, 142540).
[63] Pat. US 114666 (2003) (C.A., 2003, 138, 55976).
[64] Chodakowski J., Klis’ T., Servatowski J., Tetrahedron Letters, (2005), 46, 1963-1965.
[65] Anquetin G., Greiner J., Vierling P., Tetrahedron, (2005), 61, 8394-8404.
[66] Omura K., Uchida T., Irie R., Katsuki T., Chem. Commun., (2004), 2060- 2061.
[67] Leroux F., Hutsehenreuter T.U., Charriere C., Scopelliti R., Hartman R.W., Helv. Chim. Acta, (2003), 86, 2671-2686.
[68] Inagaki H., Tsuruoka H., Hornsby M., Lesley S.A., Spraggon G., Ellman J.A., J. Med. Chem., (2007), 50, 2693-2699.
[69] Bashore C.G., Vetelino M.G., Wirtz M.C., Brooks P.R., Frost H.N., McDermort R.E., Whritenour D.C., Ragan J.A., Rutherford J.L., Makowski T.W., Brenek S. J., Coe J.W., Org. Letters, (2006), 8, 5947-5950.
[70] Becht J.M., Ngouela S., Wagner A., Mioskowski Ch., Terahedron, (2004), 60, 6853-6858.
[71] Leroux F.R., Bonnafoux L., Heiss C., Colobert F., Lanfranchi D.A., Advanced Synth. and Catalysis, (2007), 349, 2705-2713.
[72] Milne J.E., Buchwald S.L., J. Am. Chem. Soc., (2004), 126, 13028-13032.
[73] Heis C., Leroux F., Schlosser M., Eur. J. Org. Chem., (2005), 5242-5247.
[74] Pat. US 6388146 (2002) (C.A., 1999, 131, 163453).
[75] Scott J.P., Brower S.E., Davis A.J., Brands K.M.J., Synlett, (2004), 1646-1648.
[76] Davies A.J., Scott J.P., Bishop B.C., Brands K.M., Brewer S.E., DaSilva J.O., Dormer P.G., Dolling U-H., Gibb A.D., Hammond D.C., Lieberman D.D., Palucki M., Payack J.T., J. Org. Chem., (2007), 72, 4864-4871.
[77] Pat. US 42851 (2009) (C.A., 2007, 147, 234875).
[78] Nishida J., Fujiwara Yu., Yamashita Y., Org. Letters, (2009), 11, 1813-1816.
[79] Pat. US 131482 (2009) (C.A., 2009, 150, 563826).
[80] Chen Sh., Huby N.J.S., Kong N., Moliterni J.A., Jonh A., Morales O.J., Pat. US 170920 (2009) (C.A., 2009, 151, 123990).
[81] Keum H.-W., Roh S.D., Do Y.-S., Lee J.-H., Kim Y.-B., Mol. Crystals and Liquid Crystals, (2005), 439, 189-199.
[82] Bremer M., Lietrau L., New J. Chem., (2005), 29, 72-74.
[83] Pat. US 247585 (2007) (C.A., 2007, 147, 494408).
[84] a) Glendenning M.E., Goodby J.W., Hird M., Toyne K.J., J. Chem. Soc., P2, (2000), 27-34; b) J. Chem. Soc., P2 (1999), 481-491.
[85] Cammidge A.N., Crepy K.V.L., J. Org. Chem., (2003), 68, 6832-6835. [86] Popov I., Do H-Q., Daugulis O., J. Org. Chem., (2009), 74, 8309-8313.
[87] Coe P.L., Rees A.J., J. Fluor. Chem., (2000), 101, 45-60.
[88] Do H.-Qu., Daugulis O., J. Am. Chem. Soc., (2008), 130, 1128-1129.
[89] Do H.-Qu., Khaw R.M.K., Daugulis O., J. Am. Chem. Soc., (2008), 130, 15185-15192.
[90] Pat. US 76266 (2009) (C.A., 2009, 150, 329777).
[91] Do H.-Qu., Daugulis O., Chem. Commun., (2009), 6433-6435.
[92] Matsuyama N., Kitahara M., Hirano K., Satoh T., Miura M., Org. Letters, (2010), 12, 2358-2361.
[93] Susuki A., J. Organomet. Chem., (1999), 576, 147-168.
[94] Molander G.A., Biolatto B., J. Org. Chem., (2003), 68, 4302-4314.
[95] Kotsikorou E., Song Y., Chan J.M.W., Faelens S., Tovian Z., Broderick E., Bakalara N., Docampo R., Oldfield E., J. Med. Chem., (2005), 48, 6128-6139.
[96] Song Y., Oldfield E., Lin F.-Y., Yin F, Mikkamala D., Dushyant C., Cao R., Hensler M., Nizet V., Poveda C-A.R., Pacanowska D.G., Wang H., Morita C.T., J. Med. Chem., (2009), 52, 976-988.
[97] Kumar M., Kaur K., Sinha S., Gupta S., Palle V., Chugh A., Pat US 12116 (2009) (C.A., 2007, 146, 162865).
[98] Feuerstein M., Berthiol F., Doucet H., Santelli M., Synlett, (2002), 1807-1810.
[99] Kulmaelae T., Kuuloja N., Franzen R.X., Rissanen R.K., Europ. J. Org. Chem., (2008), 4019-4024.
[100] Guo M., Zhang Q., Tetrahedron Letters, (2009), 50, 1965-1968.
[101] Duplantier A.J., Kraus K.G., Lu J., Noha M., Rogers B.N., Zhang L., Efremov I., Candler J., O’ Sillivan T.J., Ganong A.H., Hanks A.N., Lazzaro Jr. J.T., McCarthy S.A., Siuciak J.A., Doran A.C., Haas J.A., Spracklin D.K., Bioorg. Med. Chem. Letters, (2009), 19, 2524-2529.
[102] Pat. EP 1842854 (2007) (C.A., 2007, 147, 427511); Pat. WO 148829 (2008) (C.A., 2009, 150, 44027); Pat. US 200920 (2009) (C.A., 2007, 146, 430964).
[103] Pat. WO 11447 (2009) (C.A., 2009, 150, 121792).
[104] Pat. US 163508 (2009) (C.A., 2009, 150, 447978). 109
[105] Pat. WO 112854 (2009) (C.A., 2009, 151, 392217).
[106] Pat. WO 17664 (2009) (C.A., 2009, 150, 191528); Pat. WO 10150 (2010) (C.A., 2010, 152, 215298).
[107] Pat. US 203705 (2009) (C.A., 2009, 151, 245649).
[108] Pat. WO 155388 (2009) (C.A., 2010, 152, 97501); Pat. WO 155389 (2009) (C.A., 2010, 152, 97472).
[109] Pat. WO 102460 (2009) (C.A., 2009, 151, 289162).
[110] Pat. WO 153285 (2009) (C.A., 2010, 152, 97447).
[111] Ce G., Guo H., He J., Wang F., Zou D., J. Organomet. Chem., (2009), 694, 3050-3057.
[112] Lumeras W., Caturla F., Vidal L., Esteve C., Vidal B., Balaque C., Orellana A., Godessard N., Dominguez M., Roca R., Huerta J.M., J. Med. Chem., (2009), 52, 5531-5545.
[113] Imbriglio J.E., Chang S., Liang R., Raghavan S., Schmidt D., Smenton A., Tria S., Schrader T.O., Jung J.K., Esser C., Taggart A.K.P., Cheng K., Carballo-Jane E., Waters M.G., Tata J.P., Colletti S.L., Bioorg. Med. Chem. Letters, (2009), 19, 2121-2124.
[114] Matin A., Gavandl N., Kim M.S., Yang N.X., Salam N.K., Hanzahan J-R., Roubin R.H., Hibbs D.E., J. Med. Chem., (2009), 52, 6835-6850.
[115] Pat. WO 26657 (2009) (C.A., 2009, 150, 306367).
[116] Hagdar S.N., Comery T.A., Dunlop J., Ghiron Ch., Bettinetti L., Bochmann H., LaPosa S., Micco I., Pollastrini M., Quinn J., Roncarati R., Scali C., Valacchi M., Varrone M., Zanaletti R., Bioorg. Med. Chem., (2009), 17, 5247-5258.
[117] Lee S., Yi K.Y., Youn S.J., Lee B.A., Yoo S., Bioorg. Med. Chem. Letters, (2009), 19, 1329-1331.
[118] Pat. WO 52341 (2009) (C.A., 2009, 150, 472421).
[119] Blake T.D., Hamper B.C., Huang W., James R., Moon J.B., Neel B.E., Olson K.L., Pelc M.J., Schneitzer B.A., Thorarensen A., Trujillo J.I., Turner S.R., Pat. US 146569 (2008) (C.A., 2008, 149, 79492).
[120] Pat. US 170907 (2009) (C.A., 2007, 146, 62449).
[121] Burns M.J., Fairlamb I.J.S., Kapdi A.R., Sehnal P., Taylor R.J.K., Org. Letters, (2007), 9 , 5397-5400; Fairlamb I.J.S., Sehnal P., Taylor R.J.K., Synthesis, (2009), 508-510.
[122] Pat. EP 2123623 (2009) (C.A., 2008, 149, 235600). 110
[123] a) Pat WO 120789 (2009) (C.A., 2009, 151, 403339); b) Pat. EP 2116522 (2009) (C.A., 2008, 149, 343403).
[124] Li G., Stamford A.W., Huang Y., Cheng K.Ch., Cook J., Farley C., Gao J., Ghibaudi L., Greenlee W.J., Guzzi M., van Heek M., Hwa J.J., Kelly J., Mullins D., Parker E.M., Wainhaus S., Zhang X., Bioorg. Med. Chem. Letters, (2008), 18, 1146- 1150.
[125] Bouillot A.M., Dodic N., Gellibert F.J., Mirguet O., Pat. WO 15652 (2010) (C.A., 2010, 152, 238948).
[126] Demir A.S., Findik H., Saygili N., Subari N.T., Tetrahedron, (2010), 66, 1308-1312.
[127] Dong Ch.Ch., Styring P., Goodby J.W., Chan L.K.M., J. Mater. Chem., (1999), 9, 1669-1678.
[128] Yelamaggad C.V., Mathews M., Hizemath U.S., Rao D.S., Prasad S.K., Tetrahedron Letters, (2005), 46, 2623-2626.
[129] Deeg O., Bauerle P., Org. and Biomol. Chem., (2003), 1, 1609-1624.
[130] Pat. WO 52722 (2006) (C.A., 2006, 144, 488936).
[131] Tomson S.A., Banker P., Bickett D.M., Boucheron J.A., Carter H.L., Clancy D.C., Cooper J.P., Dickerson S.H., Garrido D.M., Nolte R.T., Peat A.J., Sheckler L.R., Sparks S.M., Tavares F.X., Wang L., Wang T.Y., Weiel J.E., Bioorg. Med. Chem. Letters, (2009), 19, 1177-1182.
[132] Wood W.J.L., Patterson A.W., Tsuruoka H., Jain R.K., Ellman J.A., J. Am. Chem. Soc., (2005), 127, 15521-15527.
[133] Pat. WO 77385 (2009) (C.A., 2009, 151, 77775).
[134] Liu P., Zhou L., Li X., He R., J. Organomet. Chem., (2009), 694, 2290-2294.
[135] Bey E., Marchais-Oberwinkler S., Negri M., Kruchten P., Oster A., Kllin T., Spadaro A., Werth R., Frotscher M., Birk B., Hartmann R.W., J. Med. Chem., (2009), 52, 6724-6743.
[136] Pat. WO 45383 (2009) (C.A., 2009, 150, 396692).
[137] Pat. US 258866 (2009) (C.A., 2008, 148, 17746).
[138] Jaroch S., Hölscher P., Rehwinkel H., Sülzle D., Burton G., Hillmann M., McDonald F.M., Bioorg. Med. Chem. Letters, (2002), 12, 2561-2564; Jaroch S., Rehwinkel H., Hölscher P., Sülzle D., Burton G., Hillmann M., McDonald F.M., Miklautz H, Bioorg. Med. Chem. Letters, (2004), 14, 743-746.
[139] Zhou L., Du X., He R., Ci Z., Bao M., Tetrahedron Letters, (2009), 50, 406- 408.
[140] Stencel L.M., Kormos C.M., Avery K.S., Leedbeater N.E., Org. Biomol. Chem., (2009), 7, 2452-2457.
[141] Pat. US 239748 (2009) (C.A., 2007, 146, 121670).
[142] Pat. EP 2011788 (2009) (C.A., 2007, 147, 502372).
[143] Barberousse V., Bondoux M., Tomas D., Peyrou V., Pat. US 293768 (2008) (C.A., 2006, 145, 377529); Bondoux M., Mignon L., Ou K., Renaut P., Tomas D., Barberousse V., Tetrahedron Letters, (2009), 50, 3872-3876.
[144] Eidenschink R., Kretzchmann H., Pat. US 131689 (2009) (C.A., 2007, 147, 200299).
[145] Pat. WO 91884 (2009) (C.A., 2009, 151, 208832).
[146] Pat. US 29935 (2010) (C.A., 2008, 148, 403569).
[147] Lugo B., Allbutt B., Beaumont D., Butt U., James A.R., Synlett, (2009), 675- 680.
[148] Taugerbeck A., Montenegro E., Manabe A., Plach H., Pat US 247620 (2009) (C.A., 2007, 147, 177293).
[149] Hird M., Goodby J.W., Gough N., Toyne K.J., J. Mater. Chem., (2001), 11, 2732-2742.
[150] Raghavan S., Schmidt D.R., Darby R., Steven S.L., Abi- Smenton A.L., Pat. US 62269 (2009) (C.A., 2007, 147, 277179).
[151] Korenaga T., Kosaki T., Fukumura R., Ema T., Sakai T., Org. Letters, (2005), 7, 4915-4917.
[152] a) Frohn H-J., Adonin N. Yu., Bardin V.V., Starichenko V.F., J. Fluor. Chem., (2003), 122, 195-199; b) Tetrahedron Letters, (2002), 43, 8111-8114; c) J. Fluor. Chem., (2002), 117, 115-120.
[153] Pat. WO 109743 (2009) (C.A., 2009, 151, 337021).
[154] Levi M.D., Gofer Y., Cherkinsky M., Birsa M.L., Aurbach D., Berlin A., Phys. Chem. Chem. Phys., (2003), 5, 2886-2893.
[155] Mosrin M., Knochel P., Org. Letters, (2009), 11, 1837-1840.
[156] Pat. EP 987238 (2000) (C.A., 1998, 129, 223634).
[157] Wunderlich S.H., Knochel P., Angew. Chem., Int. Ed.Engl., (2009), 48, 1501- 1504.
[158] Bromidge S.M., Dabts S., Davies D.T., Davies S., Duckworth D.M., Forbes I.T., Gaster L.M., Ham P., Jones G.E., King F.D., Mulholand K.R., Saunders D.V., Wyman P.A., Blaney F.E., Clarke S.E., Blackburn T.P., Holland V., Kennett G.A., Lightower S., Middlemiss D.N., Trail B., Rilly G.J., Wood M.D., J. Med. Chem., (2000), 43, 1123-1134. [158a] Pat. WO 14268 (2009), (C.A., (2009), 150, 213983).
[159] a) Hatakeyama T., Hashimoto S., Ishizuka K., Nakamura M., J. Am. Chem. Soc., (2009), 131, 11949-11963; b) Hatakeyama T., Nakamura M., J. Am. Chem. Soc., (2007), 129, 9844-9845.
[160] Mayer M., Czaplik W.M., Jacobi von Wangelin A., Synlett, (2009), 2919- 2923.
[161] Ok H.O., Reigle L.B., Candelore M.R., Cascieri M.A., Colwell L.F., Deng L., Feeney W.P., Forrest M.J., Hom G.J., MacIntyre D.E., Stader C.D., Tota L., Wang P., Wyvratt M.J., Fisher M.H., Weber A.E., Bioorg. Med. Chem. Letters, (2000), 10, 1531-1534.
[162] Hatakeyama T., Kondo Y., Fujiwara Yu-J., Takaya H., Nakamura M., Ito Sh., Nakamura E., Chem. Commun., (2009), 1216-1218.
[163] Van Huis Ch.A., Casimiro-Garcia A., Bigge C.F., Cody W.L., Dudley D.A., Filipski K-J., Heemstra R.J., Kohrt J.T., Leadley Jr. R.J., Narasimhan L.S., McClanahan T., Mochalkin I., Pamment M., Thomas P.T., Sahasrabudhe V., Schaum R.P., Edmunds J.J., J. Bioorg. Med. Chem., (2009), 17, 2501-2511.
[164] Pat. WO 13126 (2009) (C.A., 2009, 150, 191522); Orsini P., Menichin-Cheri M., Vanotti E., Panzeri A., Tetrahedron Letters, (2009), 50, 3098-3100.
[165] Pat. WO 70869 (2009) (C.A., 2009, 151, 33423).
[166] Davies A.J., Scott J.P., Bishop B.C., Brands K.M., Brewer S.E., DaSilva J.O., Dormer P.G., Dolling U-H., Gibb A.D., Hammond D.C., Lieberman D.D., Palucki M., Payack J.T., J. Org. Chem., (2007), 72, 4864-4871.
[167] Pat. WO 140128 (2009) (C.A., 2009, 151, 571119).
[168] Abarbri M., Dehmel F., Knochel P., Tetrahedron Letters, (1999), 40, 7449- 7454.
[169]. ЗАО НПО «Пим-Инвест». Синтезы фторорганических соединений., Изд. «Тровант», Москва, (2005), c. 123, 124.
[170] Scheuermann G.M., Steurer P., Muelhaupt R., Rumi L., Bannwarth W., J. Am. Chem. Soc., (2009), 131, 8262-8270.
[171] Zhang X., Mu F., Robinson B., Wang P., Tetrahedron Letters, (2010), 51, 600-601.
[172] Audia J.E., Mergott D.J., Sheehan S.M., Watson B.M., Pat. US 275566 (2009) (C.A., 2009, 151, 528778).
[173] Pat. WO 103007 (2009) (C.A., 2009, 151, 288987).
[174] Gordeev M.F., Pat US 48305 (2009) (C.A., 2009, 150, 214368).
[175] Pulici M., Zuccotto F., Badari A., Nuvoloni S., Cervi G., Traquandi G., Biondaro S., Trifiro P., Marchionni C., Modugno M., Pat. WO 10154 (2010) (C.A., (2010), 152, 215278).
[176] Liu J., Jian T., Guo L., Atanasova T., Nargund R.P., Tetrahedron Letters, (2009), 50. 5228-5230.
[177] Heiss C., Schlosser M., Eur. J. Org. Chem., (2003), 447-451.
[178] Morgan B.P., Galdamez G.A., Gilliard Jr. R.J., Smith R.C., Dalton Trans., (2009), 2020-2028.
[179] a) Organ M.G., Calimsiz S., Sayah M., Hoi K.H., Lough A.J., Angew. Chem., Int. Ed.Engl., (2009), 48, 2383-2387; b) Altenhoft G., Goddard R., Lehmann Ch.W., Glorius F., J. Am. Chem. Soc., (2004), 126, 15195-15201.
[180] Sakamoto Y., Suzuki T., Miura A., Fujikawa H., Tokito S., Taga Y., J. Am. Chem. Soc., (2000), 122, 1832-1833.
[181] Yudin A.K., Martyn L.J.P., Pandiaraju S., Zheng J., Lough A., Org. Letters, (2000), 2, 41-44.
[182] Pat. WO 111299 (2009) (C.A., 2009, 151, 358919).
[183] Burton D.J., Hartgraves G.A., J. Fluor.Chem., (2009), 130, 254-258.
[184] Виноградов А.С., Краснов В.И., Платонов В.Е., Ж. орган. хим., (2008), 44, 101-107. Vinigradov A.S., Krasnov V.I., Platonov V.E., Russ. J. Org. Chem., (2008), 44, 95-102 (English translation).
[185] Hori A., Kataoka H., Akacaka A., Okano T., Fujita M., J. Polymer Sci., part A: Polymer Chem., (2003), 41, 3478-3485.
[186] Facchetti A., Yoon M-H., Stern Ch.L., Katz H.E., Marks T.J., Angew. Chem., Int. Ed. Engl., (2003), 42, 3900-3903. 114
[187] Cho D.M., Parkin S.R., Watson M.D., Org. Letters, (2005), 7, 1067-1068.
[188] Blass B.E., Huang C.T., Kawamoto R.M., Li M., Liu S., Portlock D.E., Rennells W.M., Simmons M., Bioorg. Med. Chem. Letters, (2000), 10, 1543-1546.
[189] Ragni R., Orselli E., Kottas G.S., Omar O.H., Babudri F., Pedone A., Naso F., Farinola G., DeCola L., Chemistry – A Europ. J., (2009), 15, 136-148.
[190] Nagao I., Shimizu M., Hiyama T., Angew. Chem., Int. Ed. Engl., (2009), 48, 7573-7576.
[191] Frattarelli D., Ratner M.A., Marks T.J., Schiavo M., Facchetti A., J. Am. Chem. Soc., (2009), 131, 12595-12612.
[192] Pat. WO 106577 (2009) (C.A., 2009, 151, 313566).
[193] Pat. EP 1674452 (2006) (C.A., 2005, 142, 355051); Pat. EP 1780197 (2007) (yield 83%) (C.A., 2006, 144, 191974); Tecle H., Shao J., Li Y., Kuthe M., Kazmirski S., Penzotti J., Ding Y-H, Moshinsky D., Coli R., Jhawar N., Bora E., JaquesO’ Hagan S., Wu J., Ohren J., Bioorg. Med. Chem. Letters, (2009), 19, 226-229 (yield 65%).
[194] Wang C.H., Alluri S., Ganguly A.K., Tetrahedron Letters, (2009), 50, 1879- 1881.
[195] Matsuda Sh., Takahashi M., Monguchi D., Mori A., Synlett, (2009), 1941- 1944.
[196] Huang Ch., Gevorgyan V., J. Am. Chem. Soc., (2009), 131, 10844-10845.
[197] Geramita K., McBee J., Tilley T.D., J. Org. Chem., (2009), 74, 820-829.
[198] Marchetti F., Pampaloni G., Passarelli V., Masi F., Sommazzi A., Spera S., J. Fluor. Chem., (2009), 130, 341-347.
[199] Pat. WO 117097 (2009) (C.A., 2009, 151, 403303).
[200] Baumann K., Flohr A., Goetschi E., Jacobsen H., Jolidon S., Luebbers T., Pat. US 181965 (2009) (C.A., 2009, 151, 148313).
[201] Oswald C.L., Peterson J.A., Lam H.W., Org. Letters, (2009), 11, 4504-4507.
[202] Mariaca R., Labat G., Behrnd N-R., Bonin M., Helbling F., Eggli P., Couderc G., Hulliger J., Neels A., Stoeckli-Evans H., J. Fluor. Chem., (2009), 130, 175-196.
[203] Jayanth T.T., Zhang L., Johnson T.S., Malinakova H.C., Org. Letters, (2009), 11, 815-818.
[204] Takayuki T., Yuhei Y., Masashi K., Toshiaki M., Org. Letters. (2009), 11, 1043-1045.
[205] Martin R.E., Wytko J.A., Diederich F., Helv. Chim. Acta, (1999), 82, 1470- 1479.
[206] Rosenblum S.B., Huynh T., Afonso A., Davis Jr. H.R., Tetrahedron, (2000), 56, 5735-5742.
[207] Pat. US 42841 (2009) (C.A., 2009, 150, 168179).
[208] Lledo A., Restorp P., Rebeck Jr. J., J. Am. Chem. Soc., (2009), 131, 2440- 2444.
[209] a) Isaac M., Slassi A., Silva K.D., Xin T., Tetrahedron Letters, (2001), 42, 2957-2960; b) Ying M., Smentek M.G., Day C.S., Welker M.E., Ma R., Torti S.V., Letters in Org. Chem., (2009), 6, 242-251.
[210] Matsnev A., Noritake S., Nomura Y., Tokunaga E., Nakamura S., Shibata N., Angew. Chem., Int. Ed. Engl., (2010), 49, 572-576.
[211]. Biffis A., Scattolin E., Ravasio N., Zaccheria F., Tetrahedron Letters, (2007), 48, 8761-8764; Luque R., MacQuarrie D.J., Org. Biomol. Chem., (2009), 7, 1627- 1632; Johnson S.A., Liu F-Q., Suh M.C., Zuercher S, Haufe M., Mao S.S.H., Tilley T.D., J. Am. Chem. Soc., (2003), 125, 4199-4211; Zeidan T.A., Kovalenko S.V., Monoharan M., Clark R.J., Ghiviriga J., Alabugin I.V., J. Am. Chem. Soc., (2005), 127, 4270-4285; Smith C.E., Smith P.S., Thomas R.L., Robins E.G., Collings J.C., Dai Ch., Scott A.J., Borwick S., Batsanov A.S., Watt S., Clark C.J., VineyCh., Howard J.A.K., Clegg W., Marder T.B., J. Mater. Chem., (2004), 14, 413-420.
[212] a) Ojima I. Fluorine in Medicinal Chemistry and Chemical Biology, J. Wiley Publication , Willy-Blackwell, (2009), p. 299, 301; b) Benmansour H., Chambers R.D., Sandford G., Yufit D.S., Hovard J.A.K., Arkivoc, (2007), (XI), 46-55.
[213] Amii H., Uneyama K., Chem. Revs., (2009), 109, 2119-2183.
[214] Deck P.A., Kroll C.E., Hollis W.G., Fronczek F.R., J. Organomet. Chem., (2001), 637-639, 107-115.
[215] Deck P.A., McCauley B., Slebodnick C., J. Organomet. Chem., (2006), 691, 1973-1983.
[216] Gurjar M., Reddy D.S., Murugaiah A., Murugaiah S., Synthesis, (2000), 1659- 1661.
[217] [212a, p. 307].
[218] Sasaki Sh., Tanabe Y., Yoshifuji M., Bull. Chem. Soc. Japan, (1999), 72, 563- 572.
[219] Morrison D.J., Trefz T.K., Piers W.E., McDonald R., Parvez M., J. Org. Chem., (2005), 70, 5309-5312.
[220] Yang Zh-Yu., Burton D.J., J. Fluor. Chem., (2000), 102, 89-103.
[221] Wang Y., Parkin S.R., Giersehner J., Watson M.D., Org. Letters, (2008), 10, 3307-3310.
[222] Wood T.K., Waren W.E., Keay B.A., Parvez M., Angew. Chem., Int. Ed. Engl., (2009), 48, 4009-4012.
[223] Nitschke J. K., Tilley T.D., J. Am. Chem. Soc., (2001), 123, 10183-10190.
[224] Zahn A., Brotschi C., Leumann C.J., Chemistry – A Europ. J., (2005), 11, 2125-2129.
[225] Pat. WO 93467 (2006) (C.A., 2006, 145, 326323).
[226] Ishihara K., Hasegawa A., Yamamoto H., Angew. Chem., Int. Ed. Engl., (2001), 40, 4077-4079.
[227] Crouch D.J., Skabata P.J., Heeney M., Culloch I., Coles S.J., Hursthouse M.B., Chem. Commun., (2005), 1465-1467.
[228] Wang Y., Parkin S.R., Watson M.D., Org. Letters, (2008), 10, 4421-4424.
[229] Deck P.A., Lane M.J., Montgomery J.K., Slebodnick C., Frouczek F.R., Organometallics, (2000), 19, 1013-1024.
[230] Caster K.C., Keck Ch. G., Walls R.D., J. Org. Chem., (2001), 66, 2932-2936.
[231] Schlosser M., Guio L., Leroux F., J. Am. Chem. Soc., (2001), 123, 3822- 3823.
[232] Coe J.W., Wirtz M.C., Bashore C.G., Candler J., Org. Letters, (2004), 6, 1589- 1592.
[233] Pat. WO 128142 (2006) (C.A., 2007, 146, 27723).
[234] Pat. US 6362365 (2002) (C.A., 2000, 132, 308136).
[235] Tang G., Yang Y.G., Wen J.X., Mol. Cryst. Liquid Cryst. Science and Technology. Sect. A: Mol. Cryst. and Liquid Cryst., (2002), 373, 17-24.
[236] Pat WO 40131 (2006) (C.A., 2006, 144, 390561).
[237] Nuritomi M., Murofushi H., Nakashima N., Bull. Chem. Soc. Japan, (2004), 77, 2121-2128.
[238] Dutta T., Woody K.B., Watson M.D., J. Am. Chem. Soc., (2008), 130, 452- 453.
[239] Yan Sh-J., Huang Ch., Zeng X-H., Huang R., Lin J., Bioorg Med. Chem. Letters, (2010), 20, 48-51. 117
[240] Mallach E., Kuhn N., Maichle-Moessmer C., Steimann M., Stroebele M., Zeller K-P., Z. Naturforsch. B. Chemical Sciences, (2009), 64, 1176-1182.
[241] Pat. US 5856557 (1999) (C.A., 1998, 128, 88679).
[242]. Bella M., Kobbelgaard S., Jørgensen K.A., J. Am. Chem. Soc., (2005), 127, 3670-3671.
[243] Kobbelgaard S., Bella M., Jørgensen K.A., J. Org. Chem., (2006), 71, 4980- 4987.
[244] Steffen A., Sladek M.J., Braun T., Neumann B., Stammler H.G., Organometallics, (2005), 24, 4057-4064.
[245] Schaub T., Backes M., Radius U., J. Am. Chem. Soc., (2006), 128, 15964- 15965.
[246] Braun T., Perutz R.N., Sladek M.I., Chem. Commun., (2001), 2254-2255.
[247] Yoshikai N., Mashima H., Nakamura E., J. Am. Chem. Soc., (2005), 127, 17978-17979.
[248] Wang J-R., Manabe K., Org. Letters, (2009), 11, 741-744.
[249] Ishikawa S., Manabe K., Synthesis, (2008), 3180-3182; Manabe K., Ishikawa S., Synthesis, (2008), 2645-2649.
[250] Braun T., Isundu J, Steffen A., Neumann B., Stammler H-G., Dalton Trans., (2006), 5118-5123.
[251] Guo H., Kong F., Kanno K., He J., Nakajima K., Takahashi T., Organometallics, (2006), 25, 2045-2048.
[252] Korn T.J., Schade M.A., Cheemala M.N., Wirth S., Guevata S.A., Cahiez G., Synthesis, (2006), 3547-3574; Korn T.J., Schade M.A., Wirth S., Knochel P., Org. Letters, (2006), 8, 725-728.
[253] Edelbach B.L., Kraft B.M., Jones W.D., J. Am. Chem. Soc., (1999), 121, 10327-10331.
[254] Miller A.O., Krasnov V.I., Peters D., Platonov V.E., Miethchen R., Tetrahedron Letters, (2000), 41, 3817-3819.
[255] Buckley H.L., Sun A.D., Love J.A., Organometallics, (2009), 28, 6622-6624; Wang T., Love J.A., Organometallics, (2008), 27, 3290-3296.
[256] Blay G., Fernandez I., Monleon A., Pedro J-R., Vila C., Org. Letters, (2009), 11, 441-444. 118
[257] Musso D.L., Cochran F.R., Kelley J.L., McLean Ed.W., Selph J.L., Rigdon G.C., Orr G. Faye, Davis R.G., Cooper B.R., Styles V.L., Thompson J.B., Hall W.R., J. Med. Chem., (2003), 46, 399-408.
[258] Tagat J.R., McCombic S.W., Nazareno D.V., Boyll C.D., Kozlowski J.A., Chackalamannil S., Josien H., Wang Ya., Zhou G., J. Org. Chem., (2002), 67, 1171- 1177.
[259] Andrus M.B., Liu J., Tetrahedron Letters, (2006), 47, 5811-5814; Moran B.N., Anderson F.P., Cloonan S., Butler N.E., Kenny P.T.M., Deverg A., Verughese S., Draper S.M., Bioorg. Med. Chem., (2009), 17, 4510-4522.
[260] Cornella J., Lu P., Larrosa I., Org. Letters, (2009), 11, 5506-5509.
[261] Shang R., Fu Y., Wang Y., Xu Q., Yu H-Z., Liu L., Angew. Chem., Int. Ed. Engl., (2009), 48, 9350-9354.
[262] Wyatt P., Hudsen A., Charmant J., Orpen A.G., Phetmung H., Org. Biomol. Chem., (2006), 4, 2218-2232.
[263] Iovel I, Mertins K., Kirschel J., Zapf A., Beller M., Angew. Chem., Int. Ed. Engl., (2005), 44, 3913-3917.
[264] Li L., Chen H., Lin Y., Synth. Commun., (2007), 37, 985-991.
Материал рекомендован к публикации членом редколлегии В.Е. Платоновым
Fluorine Notes, 2014, 92, 1-2


