Поступило в редакцию: декабрь 2013
УДК 547.562.32
Fluorine Notes, 2013, 91, 9-10
Получение пентафторфенола окислением триметил(пентафторфенил)силана
В.Э. Бойкоa,b, A.A. Тютюновa,b, В.Л. Донa,b, С.М. Игумновa,b1
a Федеральное государственное бюджетное учреждение науки Институт элементоорганических соединений им. А.Н.Несмеянова Российской академии наук, 119991, ГСП-1, Москва, В-334, ул. Вавилова, д. 28
b ЗАО НПО "ПиМ Инвест", 119334, Москва, Ленинский пр-т 47
e-mail: boykii@mail.ru
Аннотация. Разработан новый способ получения пентафторфенола, окислением триметил(пентафторфенил)силана.
Ключевые слова: пентафторфенол, гексафторбензол, триметил(пентафторфенил)силан.
Пентафторфенол и его производные находят широкое применение для создания полимеров, обладающих уникальными свойствами [1], синтеза аминокислот, пептидов и нуклеозидов, являющихся интермедиатами в синтезе противоопухолевых агентов, ингибиторов ВИЧ [2-4], в качестве компонентов металлоценовых каталитических систем для полимеризации олефинов [5].
Все известные до настоящего времени способы получения пентафторфенола основаны на реакции нуклеофильного замещения атома фтора в гексафторбензоле гидроокисью калия [6-9] или алкоголятами [10].
В настоящее время гексафторбензол не является промышленно доступным продуктом, в связи с тем, что гексахлорбензол, из которого его получали ранее, запрещен к производству, поэтому возникла необходимость в разработке принципиально нового подхода к синтезу пентафторфенола на основе доступной сырьевой базы.
В литературе описан метод получения пентафторфенола по реакции расщепления боратов перекисью водорода в щелочной среде. Основным недостатком этого метода является необходимость для получения (пентафторфенил)триметоксибората, использовать соответствующий реактив Гриньяра, что существенно усложняет масштабирование этого метода [11, 12].
Ранее нами был разработан простой и эффективный метод получения (пентафторфенил)триметилсилана из пентафторбензойной кислоты или пентафторбромбензола [13]. Поэтому нам представлялось весьма интересным изучить возможность окисления (пентафторфенил)триметилсилана в пентафторфенол.
Варьируя природу окислителя и условия проведения реакции было обнаружено, что пентафторфенол образуется с приемлемым выходом 65% только в случае использования в качестве окислителя трет-бутилпероксибензоата и проведения реакции в присутствии KF и CuCl в ДМФА. При использовании других окислителей наблюдалось образование пентафторбензола и декафторбифенила в разных соотношениях (Табл 1).
Табл 1. Влияние природы окислителя.
|
# |
перекись |
Выход % |
||
|
PhfH |
PhfOH |
PhfPhf |
||
|
1 |
PhC(O)OOtBu |
22 |
65 |
13 |
|
2 |
tBuOOH |
89 |
0 |
11 |
|
3 |
tBuOOtBu |
91 |
0 |
9 |
|
4 |
H2O2 |
100 |
0 |
0 |
|
5 |
CO(NH2)2H2O2 |
84 |
0 |
16 |
|
6 |
BaO2 |
73 |
0 |
27 |
|
7 |
MnO2 |
49 |
3 |
48 |
|
8 |
CrO3 |
72 |
0 |
28 |
|
9 |
Na2S2O8 |
100 |
0 |
0 |
Необходимо отметить, что не наблюдается окисления (пентафторфенил)триметилсилана трет-бутилпероксибензоатом в отсутствие фтористого калия или CuCl. В последнем случае основным продуктом реакции является пентафторбензол.
Схема 1
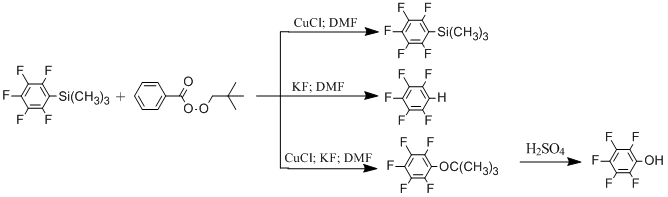
Другие соли одновалентной меди в отличие от солей двухвалентной меди также катализируют реакцию окисления.
Табл 2. Влияние природы катализатора.
|
# опыта |
Cat. |
Выход % |
||
|
PhfOH |
PhfH |
PhfPhf |
||
|
1 |
- |
0 |
100 |
0 |
|
2 |
CuCl |
65 |
22 |
13 |
|
3 |
CuBr |
52 |
30 |
18 |
|
4 |
CuI |
55 |
27 |
18 |
|
5 |
CuSO4 |
0 |
100 |
0 |
|
6 |
CH3O2SOH |
0 |
0 |
0 |
|
7 |
H2SO4 |
0 |
0 |
0 |
При изучении влияния природы растворителя на протекание данной реакции установлено, что наилучшим растворителем для данной реакции является ДМФА.
Табл. 3. Влияние природы растворителя.
|
# опыта |
Solv. |
Выход % |
||
|
PhfOH |
PhfH |
PhfPhf |
||
|
1 |
ДМФА |
65 |
22 |
13 |
|
2 |
N-метилпирролидон |
58 |
22 |
20 |
|
3 |
Тетраметилмочевина |
0 |
100 |
0 |
|
4 |
Сульфолан |
31 |
35 |
34 |
|
5 |
Ацетонитрил |
30 |
51 |
19 |
|
6 |
ДМАА |
0 |
100 |
0 |
Так же было исследовано влияние температуры проведения процесса, как оказалось, наилучший выход пентафторфенола получен при проведении процесса при 20 °С и составляет 65%, при 0°С трет-бутилпероксибензоат не вступает в реакцию, а последующее отогревание реакционной смеси до комнатной температуры приводит к неконтролируемой экзотермии до 110 °С. Если же реакцию проводить при 50-80 °С выход целевого продукта снижается до 20%.
В итоге нами был разработан новый способ получения пентафторфенола из легкодоступного триметил(пентафторфенил)силана. А так же были оптимизированы условия для проведения процесса в промышленных масштабах.
Экспериментальная часть
ЯМР 1H, 19F спектры записаны на спектрометре “Bruker AVANCE-300” при 300 и 282 MHz, соответственно, внешний стандарт CDCl3. Химические сдвиги для 1H спектров приведены относительно остаточного сигнала растворителя (δ 7.25, 4.79) и даются в м.д. относительно ТМС. Химические сдвиги спектров 19F приведены в м.д. относительно CFСl3.
В двухлитровую колбу помещают 500 мл абс. ДМФА и при интенсивном перемешивании добавляют 58г (1 моль) фтористого калия и 9.9г (0.1 моль) хлористой меди. Далее из капельной воронки к реакционной смеси охлажденной до 10°С добавляют 240г триметил(пентафторфенил)силана, реакционную смесь перемешивают 15-20минут отогревают до 20°С и затем из капельной воронки дозируют 194г третбутилпероксибензоата с такой скоростью, чтоб температура реакционной смеси не превышала 20 оС. Затем реакционную смесь разлагают равным объемом 1N HCl, отделяют органический слой, к нему добавляют ½ объема конц. HCl и кипятят до прекращения газовыделения. Затем отделяют нижний слой и перегоняют. Получают 119г пентафторфенола. Т.кип. 143 оС, Т.пл. 34-36 оС, ЯМР 1H δ: 10.5 (с, 1H,); ЯМР 19F δ: -165,1 (с, 2F 2,6), -168,3 (с, 2F 3,5), -174,7(с, 1F 4). Найдено (%): C, 39,07; H, 0,61; F, 51,69. C6HF5О. Вычислено (%): C, 39,15; H, 0,55; F, 51,61.
Список литературы
- B. Boutevin, A. Rousseau, D. Bosc; J. Polym. Sci. A, Polym.Chem., 1992, 30(7), 1279-1286;
- M. S. Ashwood, B. C. Bishop, I. F. Cottrell, K. M. Emerson, D. Hands, J. G. Ho, J. E. Lynch, Y. J. Shi, R. D. Wilson; Патент США # 7262169, 2007;
- R. P. Beckett, M. Whittaker, A. Miller, F. M. Martin; WO 9616931 1996;
- T. Sammakia, T. B. Hurley; J. Org. Chem., 2000, 65(4), 979;
- F. A.R Kaul, G. T Puchta, H. Schneider, M. Grosche, D. M. W. A Herrmann; J. Organomet. Chem., 2001, 621, 184;
- L. A.Wall, W. J.Pummer, J. E.Fearn, J. M.Antonucci; J. Res. NBS, 1963, 67A, 481-497;
- И. К. Бильдинов, Д. Ф. Мухаметшин, С. И. Коновалов, А. А. Захаров, М. М. Зиннуров; Патент РФ # 2343142 C2, 2009;
- J. M. Birchall, R. N. Haszeldine; J. Chem. Soc., 1959, 13-17;
- W. J. Pummer, L. A. Wall; Science, 1958, 127, 643;
- E.J. Forbes, R.D. Richardson, M. Stacey, J.C. Tatlow; J. Chem. Soc., 1959, 2019–2021;
- T.Korenaga, F.Kobayashi, K.Nomura, S.Nagao, T.Sakai; J. Fluorine Chem., 2007, 128, 1153–1157;
- I.Ikuyo, M.Hitoshi; Заявка на патент Японии JP 82548(A), 2005;
- V. E. Boyko, A. A. Tyutyunov, V. L. Don, S. M. Igumnov; Preparative method for obtaining of (polyfluoroaril)trimethylsilanes //Fluorine notes, 2013, 6 (91).
Статья представлена членом редколлегии С.М. Игумновым
Fluorine Notes, 2013, 91, 9-10


