Fluorine Notes, 2013, 89, 7-8
Теоретические исследования свойств структуры фтористых соединений Ванадия (III) и Молибдена (III)
Ali Rahmani
Department of Chemistry, Faculty of Science, Azad University unit Of Takestan , Takestan, Iran.
Fax: (+98) 281-3780040, e-mail: Ali_Rahmani870@yahoo.com
Аннотация. В данном теоретическом исследовании использовалась теория функциональной плотности для вычисления молекулярных структур фторидов (фтористых соединений) Ванадия (III) и Молибдена (III) VF3 и MoF3. Молекулярная геометрия, колебательные частоты, энергии и NBO-анализ (NBO-Natural bond orbital) в основном состоянии вычислены с применением DFT (B3LYP) метода с LANL2DZ. The T.S guesses были определены линейным методом синхронного перехода, при DFT implemented on Gaussian 09 program. Геометрии и обычные режимы колебаний, полученные из B3LYP вычислений, полностью согласуются с данными полученными во время экспериментов.
Ключевые слова: Теоретические исследования, фтористые соединения Ванадия (III) и Молибдена (III), Gaussian 09, VF 3 и MoF3
1. Введение
Уникальные свойства галогенидов придают необычайную реакционную способность фторидным связям, которые можно эксплуатировать в препаративной неорганической химии или катализе.
Исследование структур и свойств данных соединений, а также схожих с ними представляет большой интерес. Фтористые соединения используются много теоретически и в промышленности. Было выявлено большое количество различных данных касающихся структурных свойств соединений фтора, но они несущественны и кое-где даже противоречат друг другу. Из-за сверхвысокой электроотрицательности фтора перфторированные соединения обладают физическими свойствами, которые можно использовать в органическом синтезе и методах отделения, таких как экстракция твердой фазы.
Ванадий (III) фторид и Молибден (III) фторид (VF3 и MoF3) были использованы в структурных химических исследованиях и органическом синтезе. Теоретические расчеты использовались для получения структурных и электронных данных многих соединений, в особенности фтористых соединений. Мы применили метод DFT для оптимизации и вычисления молекулярных данных синтезированных веществ. Расчет был произведен с использованием программ Gaussian 09 AD (1993), Becke для DFT, Becke’s three-parameter exchange functional CT (1988), Lee была использована в сочетании с Lee–Yang–Parr correlation functional (B3LYP) с LANL2DZbasis set. Seppelt синтезировал соединения Ванадий (III) фторид и Молибден (III)фторид, и в этой статье мы также исследовали другие свойства данных соединений [1-11].
2. Материалы и методы
2.1 Методы вычисления
Все вычисления производились с использованием программы Gaussian 09 [12, 13]. Строения молекул полностью оптимизированы без какой-либо симметрии на всех уровнях. Оптимизированные структурные параметры использовались для вычисления колебательных частот на уровнях DFT для характеризации все стационарных точек как минимальных. Инфракрасные интенсивности (int) в км/моль выполнялись на том же уровне на соответственно полностью оптимизированных геометриях. Эти соединения и данные на них полностью согласуются с недавними работами по образованию четырех координированных промежуточных веществ.
3. Результаты
Ванадий (III) фторид и Молибден (III) фторид - VF3 и MoF3 исследовались, и оптимизация геометрии выполнялась на уровне B3LYP / LANL2DZ, она изображена на рисунке 1.
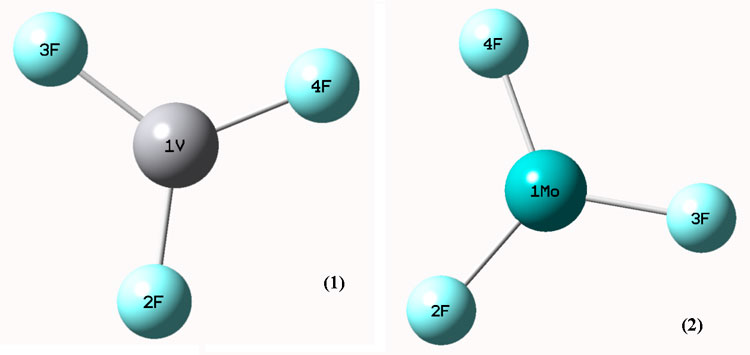
Рисунок 1. Оптимизированные геометрии VF3 и Mo3 на уровне B3LYP/LANL2DZ.
VF3, в котором F (2) связан с атомом V (1), имеет линейную F(2)–V(1) структуру с длиной связи равной 2.59 Å. F(3) связан с V(1) атомом, имеет линейную F(3)–V(1) структуру с длиной связи 2.61 Å. MoF3, в котором F(2) связан с атомом Mo(1) имеет линейную структуру F(2)– Mo(1) с длиной связи 2.06 Å. F(4) связан с атомом V(1) имеет линейную структуру F(4)–V(1) с длиной связи 2.29 Å. Отобранные расстояния между связями показаны на рисунке 1, выбранные углы - в таблице 1.
Таблица 11. Геометрические параметры, оптимизированные для (1) VF3, (2) Mo3
Как самые высокие занимаемые молекулярные орбиты(HOMO), так и самые низкие занимаемые орбиты (LUMO) являются основными орбитами, участвующими в химической стабильности. HOMO представляет собой способность отдавать электрон, LUMO как акцептор электронов представляет собой способность получать электрон, энергии HOMO и LUMO вычислены с помощью B3LYP методом LANL2DZ (Рисунок 2). Это электронное поглощение соответствует переходу от начального к первому состоянию возбуждения и, в основном, описывается возбуждением одного электрона с самой высокой занимаемой молекулярной или орбитальной позиции (LUMO).
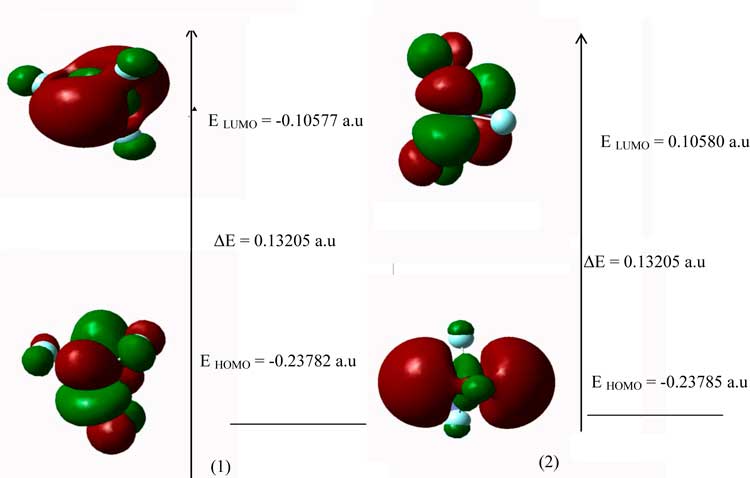
Рисунок 2. Атомные орбитали (1) VF3, (2) Mo3
Заряды атомов и порядок связей являются важными параметрами нашего исследования. Данные количества получены из анализа NBO population. Метод NBO отдает предпочтение зарядам Mulliken, так как последние обеспечивают орбитальную картинку ближе к классической структуре Льюиса. Анализ NBO затрагивающий заряды атома, порядок связей а также гибридизацию избранных связей вычисляется на уровне B3LYP/LANL2DZ.
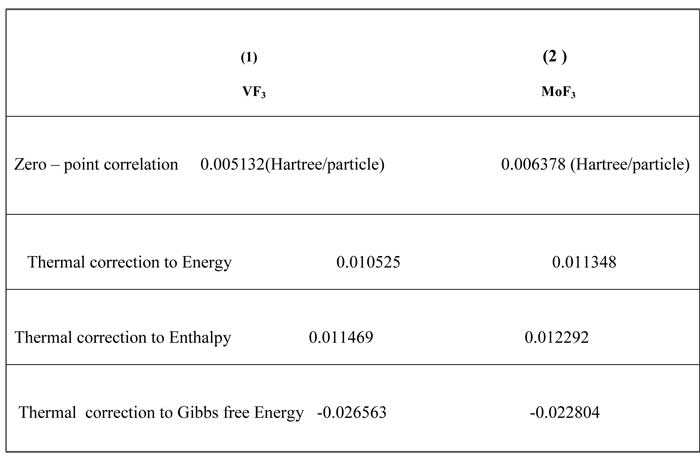
Гибридизация вычисленная по NBO для фторуглеродных соединений, VF3 и MoF3 показывает, что все соединения имеют SPX гибридизацию и неплоские структуры (Таблица 2). Анализ теории пертурбации второго порядка матрицы Фока в базе/на основе NBO для фторуглеродных соединений, VF3 и MoF3 приведен в таблице 3. Некоторые термодинамические параметры частот для (1) VF3, (2) MoF3 Zero-point Energy, correction Energy, Enthalpy lengths, Gibbs free Energy вычислены и подтверждены другими опубликованными теоретическими данными (Таблица 4).
Table2. NBO вычисленные гибридизации для (1) VF3, (2) Mo3 при B3LYP/LanL2DZ.
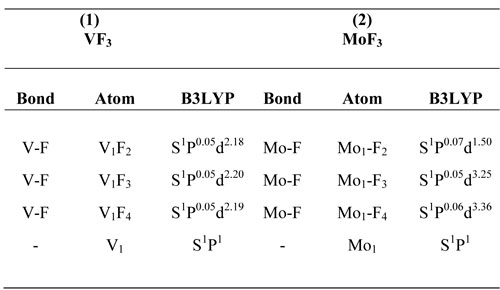
Table3. Анализ теории пертурбации второго порядка матрицы Фока для (1) VF3,
(2) Mo3
aэнергия стабилизации;
b разница энергий
между i и j NBO орбиталями;
c F(i, j) элемент матрицы Фока между i и j NBO орбиталями.
Table4. Некоторые термодинамические параметры для (1) VF3, (2) Mo3 : Zero-point Energy, correction Energy, Enthalpy lengths, Gibbs free Energy.
Данные молекулы имеют нарушенные симметрии C3v соответственно и треугольные и квадратные пирамидальные формы.
4. Обсуждение результатов
Методы теории функциональной плотности были применены для определения оптимизированных структур VF3 и MoF3. Первоначальные вычисления выполнялись на уровне DFT, использовались установки расщепленной валентности плюс поляризации LANL2DZ basis. Локальные минимумы получены с помощью полной геометрической оптимизации, они имеют все положительные частоты. Все вычисления производились с использованием компьютерной программы GAUSSIAN 09.
4.1 NBO анализ структур
(NBOs, Natural bond orbital) – это локализованные орбитали с несколькими центрами, которые описывают подобную Льюисовской связывающую молекулярную модель поведения пар электронов в оптимально компактной форме. Более точно, NBOs - это орто-нормальный набор локализованных орбиталей с «максимальной загруженностью», чьи ведущие N/2 члены (или члены N в случае открытой оболочки) дают самое точное возможное описание подобно Льюисовскому полной N-электронной плотности. Данный анализ проводился при помощи проверки всех возможных взаимодействий между «заполненными» (донорскими) Льюисовского типа NBOs и «пустыми» (акцепторными) нельюисовскими NBOs, и оценивающими их энергетическую важность теорией пертурбации второго порядка. Так как данные взаимодействия привели к передаче занятости от локализованных NBOs идеализированной Льюисовской структуры к пустым нельюисовским орбиталям (и, таким образом, к отправлениям от идеализированного описания Льюисовской структуры) они описываются как исправления «делокализации» естественной структуры Льюиса нулевого порядка. Естественные зарядки были загружены в компьютер, используя модуль орбитали естественной связи встроенный в Gaussian09. Вычисленные гибридизации NBO – это важные параметры нашего исследования. Данные количества получены из анализа популяции NBO. Последний обеспечивает орбитальную картинку ближе к классической структуре Льюиса.
4.2. Пограничная молекулярная орбиталь (Frontier molecular orbital)
Как самая высокая занимаемая молекулярная орбиталь (HOMO) так и самая низкая незанимаемая орбиталь (LUMO) – главные орбитали, участвующие в химической стабильности. HOMO демонстрирует способность быть донором электрона, LUMO как акцептор электронов представляет способность получать электрон. Как HOMO, так и LUMO энергии были вычислены с применением метода B3LYP/LANL2DZ.
Следовательно, в то время как энергия HOMO прямо связана с ионизационным потенциалом, энергию LUMO можно прямо соотнести с сродством к электрону. Разница энергий между HOMO и LUMO орбиталями называется энергетическим интервалом (energy gap), и является важной характеристикой стабильности структур.
Список литературы
- K. Seppelt, W. Sundermeyer, N-Halogensulfinylamine.Naturwissenschaften, 1969, 56: 281-282.
- Arno Schmuck, Konrad Seppelt, Sulfur Pentafluoride Cyanate F5S-O-C≡N. Angewandte Chemie International Edition in English, February 1987, Volume 26, Issue 2, pages 134–135,
- K. Seppelt, W. Sundermeyer, EineneueMethodezurHerstellung von Schwefeloxidtetrafluorid, 1971, ZAAC, 386:229-231.
- K. Seppelt, H. H.Eysel, Schwingungsspektren und Kraftkonstanten des Tetrakistrimethylsilylhydrazin, Anorg. Allg. Chem, 1971, 384:147-154.
- Bruce Sutherland, R., M. Ho.Douglas, John Huffman, C. Caulton, Kenneth G, Heterometallic Polyhydride Raft Formation: A Comparison of Syntheses usingAlkoxides of Copper and Gold. AngewandteChemie International Edition in English, 1987, 26:135-137.
- K. Seppelt, W.Sundermeyer, Über N-Halogenimidoschwefeldifluoride und N,N'-Dihalogenschwefeldiimide. Angew.Chem, 1969, 81:785-786.
- K. Seppelt, W.Sundermeyer, NeueImidoschwefeloxiddifluoride. Angew. Chem, 1970, 82:931-955.
- K. Seppelt, H.Oberhammer, Pentafluoroseleniumcyanate, F5SeOC.tplbond.N. Inorg.Chem, 1985, 24: 1227-1229.
- Becke, A.D. 1993. Density-functional thermochemistry. III. The role of exact exchange. J.Chem. Phys, 98: 5648-5652.
- Sundaraganesan, N.,S. Ilakiamani,B.Dominic Joshua, Vibrational Spectroscopyinvestigation using ab initio and density functional theory analysis on the structure of 3, 4-dimethylbenzaldehyde.Spectrochimica Acta Part A, 2007, 68:680-687.
- K. Seppelt, W.Sundermeyer, 1970. NotizuberMetalltris(trimethylsilyl)hydrazide, Chem.Ber, 103: 3939-3941.
- A. Roland, K. Seppelt, W. Sundermeyer, 19F-Kernresonanzen an Imidoschwefeloxiddifluoriden, Z. anorg.allg. Chem, (1972), 393, 149-151.
- K. Seppelt, Arsenpentachlorid, Angew. Chem. (1976), 88, 410-411.
Материал рекомендован к публикации членом редколлегии С.М. Игумновым
Fluorine Notes, 2013, 89, 7-8


