Поступило в редакцию: июль 2011
Fluorine Notes, 2013, 87, 1-2
Проявления сложной структуры потенциальных поверхностей ион-радикалов галогенаренов в спектральных (ЭПР) и химических свойствах
Л. Н. Щеголева, И. В. Береговая
Учреждение Российской академии наук Новосибирский институт органической химии им. Н.Н.Ворожцова
Сибирского отделения РАН, 630090, Новосибирск, пр. Лаврентьева, 9.
E-mail: sln@nioch.nsc.ru,
ivb@nioch.nsc.ru
Аннотация. Представлены результаты систематических квантовохимических исследований электронного и пространственного строения ион-радикалов полифтор- и некоторых хлорсодержащих соединений бензольного ряда. Рассмотрены причины и закономерности возникновения структурных искажений и их проявления в спектрах ЭПР. Исследована взаимосвязь между строением поверхностей потенциальной энергии анион-радикалов галогенаренов и их способностью к отщеплению галогенид-иона, детально изучен механизм реакции фрагментации.
Ключевые слова: Спектры ЭПР, галогенарены, отщепление галогенид-иона, квантовохимические исследования, ион-радикалы
Оглавление
I. Введение
II. Эволюция представлений о строении ион-радикалов фторароматических молекул
III. Методика расчетов потенциальных поверхностей
IV. Адиабатические поверхности потенциальной
энергии ион-радикалов бензола и гексафторбензола
V. Потенциальные поверхности ион-радикалов
частично фторированных бензолов
VI. Проявления структурной нежесткости анион-радикалов
полифторароматических соединений в спектрах ЭПР
VII. Структурные искажения и механизм
мономолекулярного распада анион-радикалов галогенаренов
I. Введение
В современной органической химии все большее внимание уделяется детальному изучению промежуточных частиц. Их идентификация, знание строения и свойств позволяют судить об элементарных стадиях и механизмах химических процессов. Многие химические и электрохимические превращения ароматических и ненасыщенных соединений, а также биологические процессы включают промежуточное образование ион-радикалов. Катион-радикалы обнаруживаются в условиях реакций ароматического электрофильного замещения [1], анион-радикалы являются ключевыми интермедиатами нуклеофильного замещения по механизму SRN1[2], участвуют в механизмах образования реактивов Гриньяра [3] и повреждения ДНК [4].
Прогресс в исследованиях ион-радикальных интермедиатов в значительной степени связан с совершенствованием экспериментальных физических методов, в особенности ЭПР, включая способы генерирования и стабилизации короткоживущих ион-радикалов в низкотемпературных матрицах, а также метод оптического детектирования ион-радикальных пар (ОД ЭПР) в растворах [5]. Не менее важную роль играют и квантовохимические исследования. Современное развитие неэмпирических методов квантовой химии и их программная реализация предоставляют возможности как для теоретической интерпретации результатов экспериментов, так и для получения независимой информации о строении и свойствах труднодоступных для экспериментальных исследований короткоживущих интермедиатов и переходных состояний химических реакций.
II. Эволюция представлений о строении ион-радикалов фторароматических молекул
Особый интерес к строению и стабильности ион-радикалов фторароматических соединений вызван их значительной ролью в химии фтораренов [6], которая существенно отличается от химии нефторированных аналогов и приводит к получению новых практически ценных продуктов – биологически активных соединений, лекарственных препаратов, полимерных материалов и др. [7-12].
Катион-радикалы целого ряда полифторароматических соединений впервые были генерированы в конце 60-х годов в Лаборатории галоидных соединений НИОХ [13,14]. Эти частицы оказались достаточно стабильными, что позволило зарегистрировать их ЭПР, УФ и ИК спектры. Интерпретация спектральных данных потребовала привлечения квантовохимических расчетов, что и послужило началом теоретических исследований ион-радикалов полифторароматических соединений в НИОХ.
Первые расчеты проводили в π-электронном приближении, используя методы Хюккеля, МакЛачлана, Паризера-Парра-Попла и геометрические параметры нейтральных предшественников. И даже эти простые расчеты позволили расшифровать весьма сложные спектры ЭПР катион-радикалов гептафторнафталинов с семью неэквивалентными ядрами 19F [15]. Позднее стали доступны методы, учитывающие все валентные электроны (CNDO/2, CNDO/S, INDO). Результаты, полученные при интерпретации констант СТВ и спектров поглощения полифторароматических катион-радикалов [16,17] полностью соответствовали сложившимся в мировой литературе представлениям об ион-радикалах ароматических молекул как о плоских π-радикалах [18,19]. Константы СТВ в спектрах ЭПР таких радикалов обусловлены исключительно спиновой поляризацией и как правило не превышают 10 Гс для ядер 1H и 50 Гс для 19F.
Анион-радикалы полифторароматических молекул гораздо менее стабильны, чем катионы. Впервые зафиксировать их удалось в 70-х годах Вильямсу и сотрудникам [20,21] благодаря развитию методов матричной изоляции при низких температурах. Неожиданно большие величины констант aF (~100-200 Гс), обнаруженные для анион-радикалов гекса- и пентафторбензола, явно не соответствовали представлению о плоской π-радикальной структуре. Проблема происхождения аномально больших величин aF в анион-радикалах фторированных бензолов интенсивно обсуждалась в литературе на протяжении почти 10 лет, при этом высказывались две основные гипотезы – плоские σ-радикалы и искаженные неплоские структуры. Первая из них основана на предположении о пересечении уровней π и σ состояний при замене атомов водорода фторами (из данных фотоэлектронной спектроскопии известно [22], что по мере накопления атомов фтора σ-уровни понижаются быстрее, чем π). Однако по данным спектроскопии электронного захвата [23], в газовой фазе молекулы полифторбензолов захватывают электрон на π уровень. Кроме того, модель плоского σ-радикала не объясняет наблюдаемый характер анизотропии СТВ (A|F > A⊥F) [24], который и лежит в основе второй гипотезы. Данные разных экспериментов противоречили друг другу, а квантовохимические расчеты были слишком несовершенны для определенного вывода.
Как экспериментальные, так и теоретические работы по исследованию полифторароматических анион-радикалов велись и в НИОХ совместно с ИХКиГ. Нам удалось разрешить проблему строения этих частиц на основе квантовохимического (INDO) анализа структурной зависимости СТВ в рамках модели, основанной на вибронном (псевдо-ян-теллеровском) взаимодействии основного π и возбужденного σ состояний анион-радикала [25]. Результатом его являются внеплоскостные искажения с отклонением атомов F от плоскости кольца, при этом симметрия искажающей координаты определяется симметрией взаимодействующих состояний. Основанием для модели послужили данные INDO расчетов о сближении (а не пересечении) π и σ уровней при замене атомов H на F. Позднее эти данные подтвердились расчетами ab initio (Рис. 1). Показанная на Рис. 1 схема уровней объясняет не только происхождение внеплоскостных искажений в анионе гексафторбензола, но и плоскую структуру катиона, для которого замена атомов H на F приводит к обратному эффекту.
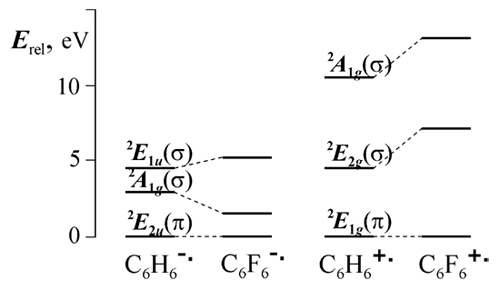
Рис. 1. Относительные уровни энергии низших электронных состояний ион-радикалов бензола и гексафторбензола (ROHF/6-31G*).
Предложенная модель позволила выявить закономерности возникновения внеплоскостных искажений и объяснить данные ЭПР для анион-радикалов широкого круга фторсодержащих производных бензола, среди которых имеются как плоские π- и σ-радикалы, так и искаженные неплоские псевдо-π- и псевдо-σ-структуры, ОЗМО которых являются линейными комбинациями π и σ орбиталей. Наличие прямого вклада s-АО фтора в ОЗМО и объясняет происхождение необычно больших величин констант СТВ с ядрами 19F при нарушении плоской структуры радикалов π-типа.
Причина внеплоскостных искажений – взаимодействие электронных состояний разной четности (π и σ) по отношению к плоскости сопряженной системы. Дальнейшие исследования в рамках вибронного подхода показали, что в ион-радикалах ненасыщенных систем, в там числе фторароматических, возможны также искажения, обусловленные взаимодействием состояний одинаковой четности [26,27]. Такие искажения носят плоскостной характер. В частности, искажения этого типа приводят к нарушению эквивалентности фрагментов в ион-радикалах симметричных молекул и локализации спиновой плотности на одном них.
Результаты исследования причин и закономерностей возникновения структурных искажений с понижением симметрии в ион-радикалах ароматических и ненасыщенных молекул обобщены в монографиях [28,29]. Эти данные привели к новым взглядам на электронное и пространственное строение ион-радикалов полифторароматических молекул. Дальнейшее развитие представлений о строении этих ионов связано с неэмпирическими исследованиями поверхностей потенциальной энергии (ППЭ), которые привели к понятию структурной нежесткости.
При искажениях с понижением симметрии потенциальная поверхность ион-радикала не является одноямной. В простейшем случае единственной невырожденной активной моды Qu на поверхности имеется два эквивалентных минимума (результат искажения вдоль ±Qu), а связывающая их седловая точка отвечает высокосимметричной структуре радикала. При участии нескольких активных координат число стационарных структур ППЭ возрастает и установление их взаимосвязей становится нетривиальной задачей, которая требует специального исследования.
Проявления сложного строения потенциальной поверхности в физико- химических свойствах ион-радикалов зависят от высоты энергетических барьеров и электронного строения системы в минимумах. Поэтому изучение свойств систем, для которых обнаруживаются структурные искажения с понижением симметрии, неразрывно связано с исследованием ППЭ с применением современных неэмпирических методов квантовой химии.
III. Методика расчетов потенциальных поверхностей
Для исследований ППЭ ион-радикалов фторароматических молекул нами развит следующий симметрийный подход [30,31]. Первоначальная оптимизация геометрии, проводится в рамках симметрии нейтральной молекулы предшественника, что отражает релаксацию образующегося ион-радикала вдоль полносимметричных мод. При найденной геометрии проводятся точечные расчеты электронных состояний, принадлежащих разным представлениям группы симметрии, и анализируются возможные в результате их вибронного взаимодействия структурные искажения. Последующая оптимизация геометрии этих состояний с сохранением симметрии нейтральной молекулы приводит к локализации стационарных структур ион-радикала, различающихся симметрией основного электронного состояния. Чтобы определить, лежит ли найденная структура на листе ППЭ основного состояния, проводятся дополнительные точечные расчеты с изменением начального приближения. Это позволяет выявить возможные пересечения термов состояний разной симметрии. Тип найденных стационарных точек ППЭ определяется путем анализа нормальных колебаний. При обнаружении мнимых колебательных частот рассматривается понижение симметрии вдоль соответствующей активной моды. Для установления взаимосвязей между стационарными точками используется метод внутренней координаты реакции (IRC). Описанный подход позволяет установить генезис и взаимосвязи стационарных структур ион-радикалов и построить полные схемы потенциальных поверхностей.
В основе проведенных исследований лежат расчеты хартри-фоковского (ХФ) уровня [32,33], необходимые для понимания природы эффектов. Для систем с открытой электронной оболочкой использовали метод ROHF, реализованный в программе GAMESS.34 В ХФ расчетах использовали стандартный базисный набор 6-31G, расширенный поляризационными d-функциями на тяжелых атомах (6-31G*) для катион-радикалов и дополненный диффузными sp-функциями на тяжелых атомах (6-31+G*) для анионов. Для учета электронной корреляции использовали методы CIS, RMP2, CASSCF. Расчеты констант СТВ проводили методом DFT с функционалом B3LYP в неограниченном по спину варианте (UB3LYP), этот метод достаточно широко применяли также для оптимизации геометрии стационарных структур ион-радикалов.
Исследование ППЭ анион-радикалов бензола и его фторпроизводных C6FnH6-n осложняется тем, что молекулы c n < 4 имеют отрицательное адиабатическое сродство к электрону [6]. При расчетах анионов таких систем расширение базисного набора диффузными функциями может приводить к получению ХФ решений, отвечающих несвязанным состояниям свободного электрона и нейтральной молекулы [35,36]. Для отбора связанных (т.е. анион-радикальных) решений применяли простейший стабилизационный метод – масштабирование показателей экспонент диффузных функций [35]. При этом показано, что ROHF/6-31+G* решения π-типа, в отличие от σ-решений, являются связанными, дополнительный электрон остается в валентной области.
IV. Адиабатические поверхности потенциальной энергии ион-радикалов бензола и гексафторбензола
Чтобы проследить изменения ППЭ, обусловленные накоплением атомов фтора, рассмотрим сначала ион-радикалы высокосимметричных (D6h) молекул бензола и гексафторбензола. Симметричная D6h ядерная конфигурация, в которой все четыре иона имеют двукратно вырожденное основное состояние (2E1g: ...(1e1g)3(1e2u)0 для катионов и 2E2u: ...(1e1g)4(1e2u)1 для анионов), неустойчива вследствие эффекта Яна-Теллера (ЭЯТ). Активная ян-теллеровская мода также вырождена и принадлежит представлению e2g. Вид адиабатической ППЭ для этого случая (ЭЯТ E×e) хорошо известен [37], это поверхность конического пересечения с тремя эквивалентными минимумами и тремя седловыми точками (Рис. 2). Стационарные структуры связаны между собой координатой псевдовращения, вдоль которой осуществляется обход точки конического пересечения. Сечение ППЭ показано на Рис. 3.
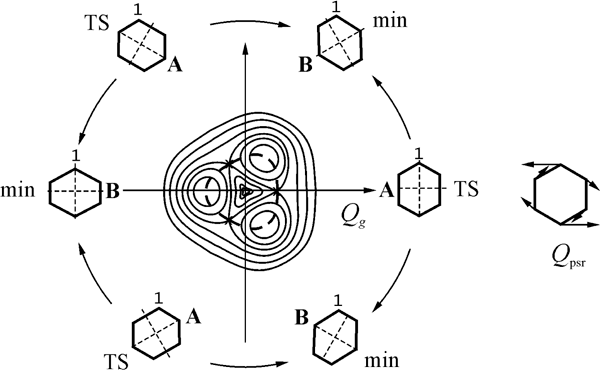
Рис. 2. Вид адиабатической ППЭ ян-теллеровских систем, обладающих осью C3, и схема псевдовращения плоских D2h структур ион-радикалов бензола и гексафторбензола. Пунктиром обозначен желоб псевдовращения, справа показана соответствующая координата.
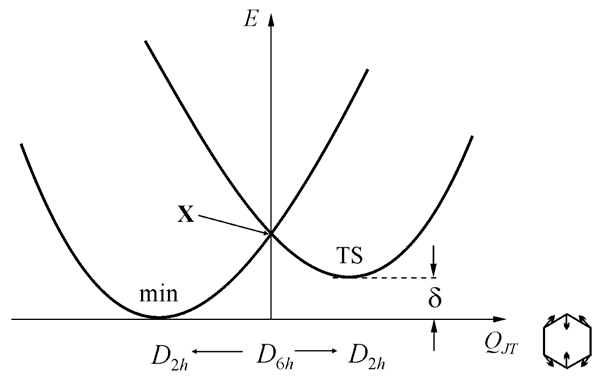
Рис. 3. Сечение ППЭ Рис. 2 вдоль полносимметричной в группе D2h координаты QJT (X–точка конического пересечения, δ – высота барьера псевдовращения).
Искажение вдоль активной координаты приводит к понижению симметрии до D2h и расщеплению вырожденных состояний. Для каждого иона возникают две D2hструктуры (искажения вдоль ±QJT), сжатая и вытянутая, различающиеся симметрией основного состояния. Далее будем обозначать эти структуры как B и A, соответственно (обозначения и вид ОЗМО показаны на Рис. 4). Одна из них отвечает минимуму, а другая – переходному состоянию псевдовращения.
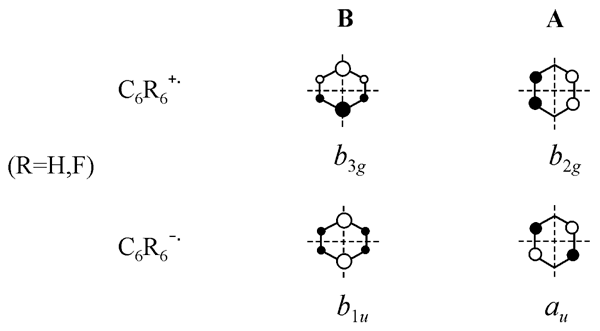
Рис. 4. Обозначения плоских D2h структур ион-радикалов бензола и гексафторбензола и вид соответствующих ОЗМО.
В случае ион-радикалов бензола и катион-радикала гексафторбензола плоские D2h структуры отвечают истинным минимумам и седловым точкам ППЭ. Однако анион-радикал гексафторбензола, как уже отмечено выше, подвержен внеплоскостным искажениям, вызванным присутствием низколежащего возбужденного σ состояния (псевдоэффект Яна-Теллера).
Последовательное понижение симметрии анион-радикала гексафторбензола вдоль активных ян-теллеровских и псевдо-ян-теллеровских мод (e2g и e2u, соответственно) рассмотрено нами на основе ROHF расчетов с учетом корреляционных поправок к энергии на уровне RMP2 [38]. Сочетание в C6F6—. ЭЯТ и ПЭЯТ приводит к удвоению числа стационарных точек потенциальной поверхности. Дополнительное понижение симметрии вдоль активных внеплоскостных мод, приводит к структурам симметрии C2v и D2. При этом каждая плоская структура порождает две зеркально симметричные неплоские. Взаимосвязи неплоских структур найдены с помощью IRC расчетов. Результирующая схема псевдовращения в C6F6—. (Рис. 5) включает шесть минимумов (структуры симметрии C2v) и шесть переходных состояний (D2 структуры), высота барьера по данным RMP2/6-31+G*//ROHF/6-31+G* расчетов составляет 0.2 ккал/моль. Энергетически эквивалентные структуры связаны между собой операцией симметрии S6, а диаметрально противоположные структуры цикла псевдовращения – операцией инверсии E*, обращающей пространственные координаты всех частиц [39]. Путь псевдовращения одновременно является путем инверсии внеплоскостного искажения.
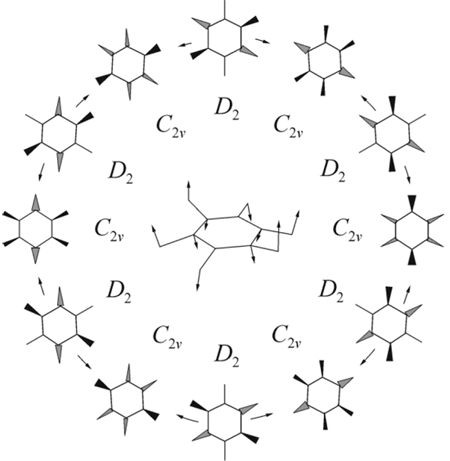
Рис. 5. Схема псевдовращения в анион-радикале C6F6—. и вид активной координаты.
Принципиальная особенность полученного цикла заключается в том, что плавное изменение геометрии при движении вдоль желоба псевдовращения сопровождается непрерывным изменением электронной волновой функции, скачок фазы, характерный для схем псевдовращения из шести плоских структур ионов C6H6+/—.[40], отсутствует. Для выяснения происхождения этой особенности поставим в соответствие каждой структуре схемы Рис. 5 порождающую ее плоскую D2h структуру, учитывая при этом, что направление внеплоскостных смещений определяется знаком плотности электронного перехода и полагая фазовый множитель неизменным (Рис. 6а).
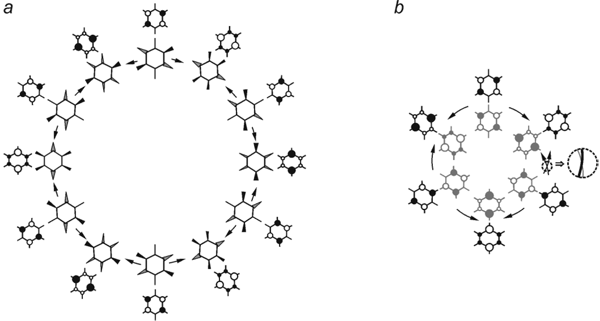
Рис 6. Соответствие искаженных и плоских структур C6F6—. (а) и схематическое изображение ленты Мебиуса – результата склеивания диаметрально противоположных плоских структур цикла псевдовращения, различающихся фазой волновой функции (b).
Построенный таким образом цикл псевдовращения включает двенадцать плоских D2h структур и, в отличие от цикла из шести структур, характеризуется непрерывным изменением электронной волновой функции (Рис. 6а). При этом диаметрально противоположные плоские структуры, как и порожденные ими неплоские, связаны операцией инверсии, но, ввиду неизменности ядерных координат при действии E*, отличаются только знаком волновой функции, т.е. физически неразличимы. Для их объединения воспользуемся топологической теоремой: [41] "склеивание каждых двух диаметрально противоположных точек одной из окружностей кольца дает ленту Мебиуса". Результат такого "склеивания" (Рис. 6b) приводит к известной схеме псевдовращения из шести плоских структур анион-радикала бензола. Разрезание ленты Мебиуса по средней линии снова дает цикл из двенадцати плоских структур, который по построению находится в однозначном соответствии со схемой псевдовращения искаженных C2v и D2 структур C6F6—. (Рис. 5), полученной из IRC расчетов. Таким образом, схемы псевдовращения в C6H6—. и C6F6—. соотносятся между собой как лента Мебиуса и результат ее разрезания по средней линии.
V. Потенциальные поверхности ион-радикалов частично фторированных бензолов
Псевдовращение в ион-радикалах бензола и гексафторбензола – результат обхода пересечения (вырождения) термов π состояний в высокосимметричной D6h ядерной конфигурации. Асимметричное замещение снимает вырождение, но атомы фтора слабо возмущают π-систему и энергетическое расщепление вырожденных в бензоле уровней граничных π-МО в молекулах низкосимметричных (D 2h, C2v) частично фторированных бензолов невелико [6]. Поэтому в их ион-радикалах имеются два близких по энергии π-состояния, в которых неспаренный электрон заселяет разные компоненты вырожденной в бензоле МО. Соответственно, для каждого иона существуют две плоские π-структуры [31,42], различающиеся симметрией электронного состояния (орбитальные изомеры), A (2A2) и B (2B1), в обозначениях группы C2v. Разность их энергий ΔEAB минимальна для ион-радикалов 1,2,3-трифторбензола, π-МО которого квазивырождены, и наиболее значительна для ионов молекул 1,4-F2C6H4 и 1,2,4,5-F4C6H2, в которых расщепление уровней вырожденных МО бензола максимально. Ввиду слабого возмущения бензольной π-системы атомами фтора, ОЗМО структур A и B ион-радикалов низкосимметричных фторбензолов близки к изображенным на Рис. 4.
Проведенный нами [31,42] на ХФ уровне расчетов анализ сечений ППЭ вдоль связывающей π-структуры полносимметричной координаты показал, что во всех случаях, кроме 1,4-F2C6H4±• и 1,2,4,5-F4C6H2±•, термы π-состояний пересекаются. По данным расчетов можно выделить три основных типа сечений, схематично изображенные на Рис. 7 (структура X отвечает точке пересечения π-термов). К первому из них, Рис. 7 а (ΔEAB<3.5 ккал/моль), кроме ян-теллеровских ионов C6F6±• и 1,3,5-F3C6H3±• относятся оба иона 1,2,3-F3C6H3±•. Другому предельному случаю, Рис. 7b (ΔEAB>17 ккал/моль), соответствуют ионы 1,4-1,4-F2C6H4±• и 1,2,4,5-F4C6H2±•, π-термы которых не пересекаются. Ионы всех остальных фторбензолов (5.5 <ΔEAB> 12 ккал/моль) принадлежат промежуточному случаю, Рис. 7c. Заметим, что тип сечения ППЭ определяется взаимным расположением атомов фтора в кольце и не зависит ни от их числа, ни от знака заряда иона.
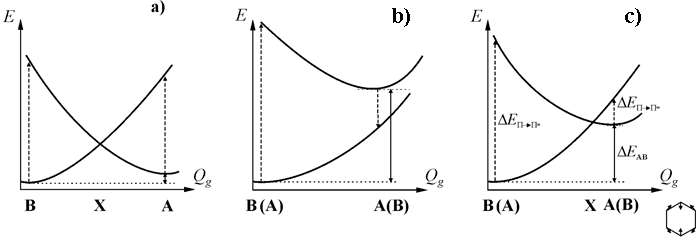
Рис. 7. Типы сечений ППЭ ион-радикалов фторбензолов вдоль координаты Qg, связывающей A и B структуры: предельные случаи малого (а) и большого (b) расщепления их уровней (ΔEAB) и промежуточный случай (c).
Таким образом, по данным ХФ расчетов потенциальные поверхности ион-радикалов большинства фторбензолов являются поверхностями конического пересечения. Обход пересечения, как и в ян-теллеровских ионах, осуществляется вдоль координаты псевдовращения, симметрия которой (b2 в группе С2v) определяется симметрией пересекающихся термов. ППЭ анион-радикалов дополнительно усложняются за счет внеплоскостных искажений, что, как и для C6F6—., можно моделировать соответствующим разрезанием мебиусовской схемы псевдовращения плоских структур.
Потенциальные поверхности ион-радикалов низкосимметричных молекул менее симметричны, а барьеры псевдовращения значительно выше, чем для ян-теллеровских систем, и варьируются от 3 до 12 ккал/моль на ХФ уровне расчетов. Учет электронной корреляции существенно влияет как на высоту барьеров, так и на число стационарных структур желоба псевдовращения [31,42]. Зависимость результатов от уровня расчетов рассмотрим на примере ион-радикалов 1,2,3-F3C6H3±• и 1,2,4,5-F4C6H2±•, сечения ППЭ которых относятся к разным предельным типам, Рис. 7а и 7b, соответственно.
Катион-радикалы фторированных бензолов имеют плоскую структуру, поэтому строение потенциальной поверхности полностью определяется пересечением термов π-состояний. В случае катиона симметричного тетрафторбензола пересечение отсутствует и на пост-ХФ уровнях расчетов, поверхность одноямна.
Для катиона 1,2,3-трифторбензола термы пересекаются, результатом обхода пересечения является псевдовращение, которое сохраняется независимо от уровня расчетов [42,43]. Учет электронной корреляции приводит к уменьшению числа стационарных структур ППЭ, которая с поправкой на энергию нулевых колебаний состоит из одного минимума и одного переходного состояния (Табл. 1). Барьер псевдовращения достаточно низок, ~1.5-2 ккал/моль (Табл. 1), что указывает на структурную нежесткость катион-радикала и возможность ее спектральных проявлений. К сожалению, в работе [44], посвященной исследованию катион-радикалов фторированных бензолов методом ЭПР, данные для 1,2,3-F3C6H3+• отсутствуют. Однако необычное строение этого катиона было отмечено Миллером, Бондибеем и сотрудниками [45,46] при исследовании спектров лазерного возбуждения и флуоресценции.
Таблица 1. Влияние уровня расчетов на форму потенциальной поверхности катион-радикала 1,2,3-F3C6H3+• (полные энергии в а.е., относительные – в ккал/моль).
|
Метод |
Структура |
Etot |
Na |
Erel |
ErelZPE |
|
ROHF/6-31G* |
C2v (2B1) |
-526.928356 |
0 |
0 |
0 |
|
|
C2v (2A2) |
-526.923255 |
1 |
3.20 |
1.05 |
|
|
Cs (2А") |
-526.925137 |
0 |
2.02 |
1.63 |
|
|
Cs (2А") |
-526.924458 |
1 |
2.45 |
0.75 |
|
CASSCF(5,5)/6-31G* |
C2v (2B1) |
-526.975333 |
1 |
0.15 |
0 |
|
|
C2v (2A2) |
-526.972963 |
1 |
1.64 |
1.40 |
|
|
Cs (2А") |
-526.975570 |
0 |
0 |
0.74 |
|
RB3LYP/6-31G* |
C2v (2B1) |
-529.356997 |
0 |
0 |
0 |
|
|
C2v (2A2) |
-529.353080 |
1 |
2.46 |
1.28 |
a Число мнимых колебательных частот
На потенциальной поверхности анион-радикала 1,2,3-F3C6H3— •, независимо от уровня расчетов, обнаруживаются четыре типа неплоских стационарных структур (Рис. 8) – два минимума (структуры Сs и С1(B)) и две седловые точки (структуры С2 и С1(A)) [Для структур симметрии C1 в скобках указан тип ОЗМО (A = a2, B = b1)], которые отвечают переходным состояниям псевдовращения [31,47,48]. Схема псевдовращения в этом анион-радикале образована двенадцатью структурами и аналогична изображенной на Рис. 5 для C6F6— •, отличаясь тем, что атомы водорода остаются в плоскости кольца, а минимумы, и также седловые точки поверхности, энергетически неэквивалентны. Барьер псевдовращения достаточно низок (Табл. 2). Таким образом, оба иона 1,2,3-трифторбензола являются структурно нежесткими.
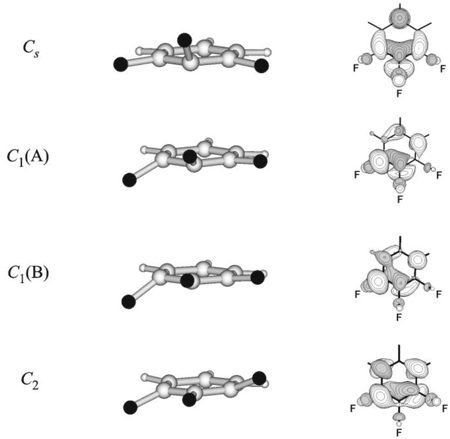
Рис. 8. Стационарные структуры ППЭ анион-радикала 1,2,3-трифторбензола и изображения соответствующих ОЗМО.
Таблица 2. Стационарные структуры анион-радикала 1,2,3-трифторбензола (полные энергии даны в а.е., относительные – в ккал/моль).
|
Метод |
Структура |
Etot |
N а |
Erel, |
ErelZPE |
|
ROHF/6-31+G* |
Cs |
-527.204096 |
0 |
0.93 |
0.92 |
|
|
C1(A) |
-527.198222 |
1 |
4.62 |
3.62 |
|
|
C1(B) |
-527.205581 |
0 |
0 |
0 |
|
|
C2 |
-527.195525 |
1 |
6.31 |
5.19 |
|
RMP2/6-31+G* |
Cs |
-528.556354 |
|
0 |
- |
|
|
C1(A) |
-528.550305 |
|
3.80 |
- |
|
|
C1(B) |
-528.550590 |
|
3.62 |
- |
|
|
C2 |
-528.549831 |
|
4.09 |
- |
|
UB3LYP/6-31+G* |
Cs |
-529.709613 |
0 |
0 |
0 |
|
|
C1(A) |
-529.705929 |
1 |
2.31 |
1.81 |
|
|
C1(B) |
-529.706811 |
0 |
1.76 |
1.68 |
|
|
C2 |
-529.703361 |
1 |
3.92 |
3.13 |
a Число мнимых колебательных частот
В случае анион-радикала 1,2,4,5-F4C6H2— • термы π-состояний плоских структур не пересекаются, но обе плоские структуры неустойчивы по отношению к внеплоскостным искажениям. Потенциальная поверхность снова имеет вид поверхности псевдовращения как результат пересечения потенциальных поверхностей неплоских структур разной симметрии. Цикл псевдовращения (Рис. 9) образован двумя эквивалентными минимумами (структуры симметрии C2v) и двумя переходными состояниями (D2 структуры). Особенностью этого аниона являются значительные внеплоскостные смещения атомов H в структурах минимальной энергии. Зеркально-симметричные C2v структуры связаны не только псевдовращением, но и прямой инверсией (через переходные состояния симметрии C2h), причем второй путь выгоднее энергетически. В отличие от барьера псевдовращения, барьер прямой инверсии при переходе к пост-ХФ методам повышается, оставаясь, тем не менее, ниже барьера псевдовращения (Табл. 3).
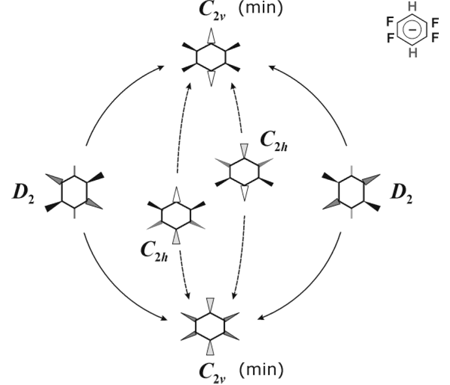
Рис. 9. Схема поверхности потенциальной энергии анион-радикала 1,2,4,5-F4C6H2— •
Таблица 3. Стационарные структуры анион-радикала 1,2,4,5-тетрафторбензола (полные энергии в а.е., относительные – в ккал/моль)
|
Метод |
Структура |
Etot |
N а |
Erel, |
ErelZPE |
|
ROHF/6-31+G* |
C2v(z) |
-626.064651 |
0 |
0 |
0 |
|
|
D2 |
-626.044396 |
2 |
12.71 |
11.72 |
|
|
C2(y) |
-626.047091 |
1 |
11.01 |
10.35 |
|
|
C2h |
-626.063785 |
1 |
0.54 |
-0.37 |
|
UB3LYP/6-31+G* |
C2v(z) |
-628.919899 |
0 |
0 |
0 |
|
|
D2 |
-628.911831 |
2 |
5.06 |
4.26 |
|
|
C2(y) |
-628.919391 |
1 |
4.71 |
4.31 |
|
|
C2h |
-628.915403 |
1 |
2.82 |
2.28 |
|
RMP2/6-31+G* |
C2v(z) |
-627.544517 |
0 |
0 |
- |
|
|
D2 |
-627.537945 |
1 |
4.12 |
- |
|
|
C2h |
-627.541180 |
1 |
2.09 |
- |
a Число мнимых колебательных частот
Рассмотренные предельные ситуации показывают, что псевдовращение в ион-радикалах низкосимметричных частично фторированных бензолов сохраняется и на пост-ХФ уровне расчетов. Все анионы этого ряда и некоторые катионы являются структурно нежесткими по отношению к координате псевдовращения.
Проведенные исследования выявили причину сходства и различий в строении ППЭ катион- и анион-радикалов фторированных бензолов.49,50 Общие их черты связаны со слабым возмущением вырожденных граничных π-МО бензола атомами фтора. Следствиями этого являются близость уровней π-состояний ион-радикалов и возможность пересечения их термов. Основная причина различий – сильное возмущение атомами фтора σ-системы бензола, что приводит к понижению уровней возбужденных σ-состояний в анионах и их повышению в катионах. Результатом является неустойчивость плоских ядерных конфигураций анион-радикалов, тогда как катионы сохраняют плоскую структуру. В анион-радикалах фторированных бензолов внеплоскостные искажения орбитально изомерных структур A и B порождают структуры, принадлежащие разным точечным группам симметрии. На примере C6H5F — • показано [49], что в этом случае псевдовращение обусловлено обходом пересечения потенциальных поверхностей неплоских ядерных конфигураций разной симметрии по трехмерной координате и потому существует независимо от пересечения π-термов плоских структур. Таким образом, анион-радикалы всех фторбензолов являются в той или иной степени структурно нежесткими.
VI. Проявления структурной нежесткости анион-радикалов полифторароматических соединений в спектрах ЭПР
Согласно изложенным выше результатам квантовохимических исследований ППЭ, анион-радикалы фторированных бензолов структурно нежестки по отношению к координате псевдовращения. Значительное перераспределение спиновой плотности в процессе псевдовращения, в котором участвуют структуры разной пространственной симметрии (см. Рис. 8), позволяет ожидать, что предсказанная расчетами структурная нежесткость будет проявляться в виде температурной зависимости сверхтонких расщеплений в спектрах ЭПР. Таким образом, возможна экспериментальная проверка полученных результатов.
Необходимое условие для этого – надежное описание констант СТВ с ядрами 19F в неплоских анион-радикалах псевдо-π-типа. Согласно результатам, полученным для анионов C6H5F — • и C5NF5— • [50] относительно высокие барьеры псевдовращения для которых (~5 и 11 ккал/моль, соответственно) позволяют считать, что спектры практически не зависят от температуры, экспериментальные величины aF хорошо воспроизводятся расчетами методом UB3LYP/6-31+G* (Табл. 4). Учитывая чувствительность величин aF к структурным параметрам, можно говорить также об адекватном описании геометрии анион-радикалов на этом уровне расчетов.
Таблица 4. Рассчитанные (UB3LYP/6-31+G*) для структур минимальной энергии (Cs) C6H5F — • и C5NF5— • и экспериментальные [51-53] константы aF (Гс).
|
Анион-радикал |
Ядро |
Расчет |
Эксперимент |
|
|
матрица |
раствор |
|||
|
C6F5H— • |
19F (о-) |
54.2 |
45.5 |
48.7 |
|
|
19F (м-) |
112.6 |
101.0 |
107.0 |
|
|
19F (п-) |
268.8 |
279.0 |
295.0 |
|
C5NF5— • |
19F (α-) |
19.5 |
17.4 |
|
|
|
19F (β-) |
69.0 |
61.2 |
|
|
|
19F (γ-) |
255.5 |
249.0 |
|
|
|
14N |
3.4 |
3.6 |
|
В качестве объекта для экспериментального исследования был выбран не описанный ранее в литературе анион-радикал 1,2,3-трифторбензола, для которого высота барьера псевдовращения минимальна в ряду анионов фторированных бензолов. Специально проведенные Вьюшковой, Багрянским и Молиным (ИХКиГ СО РАН) ОД ЭПР эксперименты показали, что спектр 1,2,3-F3C6H3 — • зависит от температуры [47,48]. При повышении температуры происходит заметный сдвиг линий спектра без их уширения, что характерно для быстрого спектрального обмена [54].
Для интерпретации наблюдаемых изменений в спектре мы использовали результаты IRC расчетов методом UB3LYP/6-31+G* (Рис. 10). Усреднение рассчитанных вдоль желоба псевдовращения величин констант СТВ с ядрами 19F в рамках классического распределения Больцмана хорошо воспроизводит температурную зависимость спектра ЭПР, наблюдаемую в интервале 243-325 K (Рис. 11). Таким образом, получено экспериментальное подтверждение структурной нежесткости аниона 1,2,3-F3C6H3 — • по отношению к координате псевдовращения.
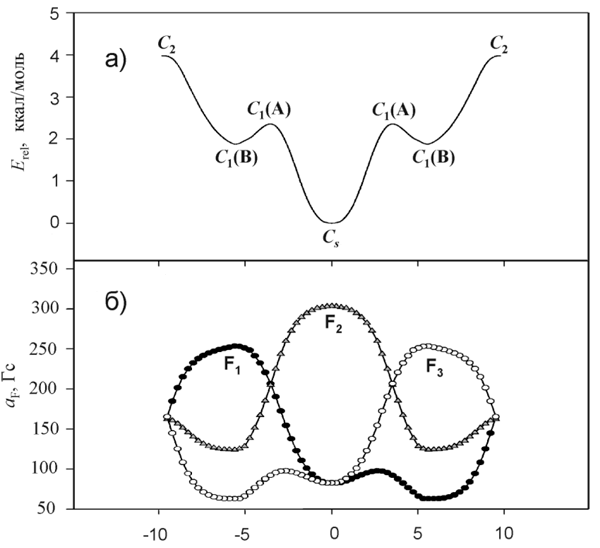
Рис. 10. Изменение энергии (а) и констант СТВ с ядрами 19F (б) анион-радикала
1,2,3-F3C6H3 — • при движении вдоль
координаты псевдовращения, UB3LYP/6-31+G* (показана половина желоба).
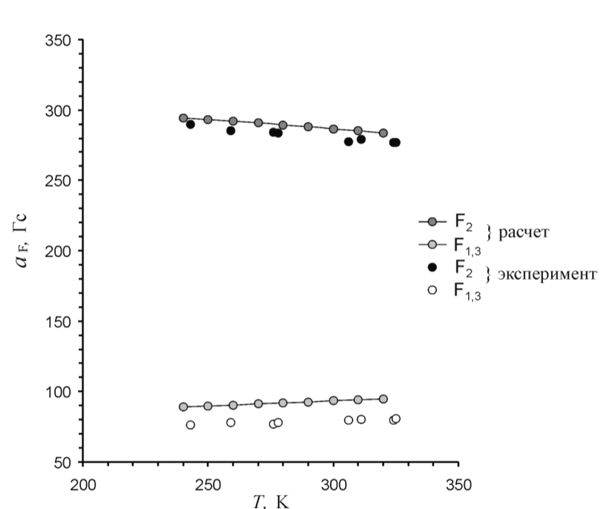
Рис. 11. Температурная зависимость констант СТВ с ядрами 19F в анион-радикале 1,2,3-F3C6H3 — • по данным ОД ЭПР и расчетов в классической модели.
В ходе дальнейшей совместной работы с экспериментаторами из ИХКиГ было показано, что структурная нежесткость не является исключительным свойством анион-радикалов бензольного ряда. Пример структурно нежесткой системы, спектр ЭПР которой не зависит от температуры, представляет недавно зафиксированный методом ОД ЭПР анион-радикал перфторбифенила, (C6F5)2— • [55]. Экспериментальные величины констант aF, 72 (2F), 26 (4F) и 18.6 (4F) Гс, однозначно указывают на отклонения атомов фтора от плоскостей колец. Учитывая, что НСМО молекулы перфторбифенила является симметричной комбинацией b1 орбиталей колец, можно было ожидать, что при захвате электрона внеплоскостное отклонение наиболее существенно для пара-атомов F, понижение симметрии при этом D2 → C2. Однако неплоские структуры симметрии C2 не позволяют объяснить наблюдаемые величины aF.
Выполненное для интерпретации данных ОД ЭПР исследование поверхности потенциальной энергии показало [55,56], что в (C6F5)2— • сочетаются все три типа исследованных нами ранее структурных искажений – обход конического пересечения, внеплоскостные отклонения атомов фтора и нарушение эквивалентности фенильных колец. Причина последнего – существенное нарушение копланарности колец в результате стерического взаимодействия орто-атомов F.
Минимуму энергии анион-радикала (C6F5)2— • соответствует полностью асимметричная C1 структура с повернутыми друг относительно друга на ~45° неэквивалентными кольцами и в разной степени отклоненными от их плоскостей атомами фтора (Рис. 12). Плотность неспаренного электрона локализована в кольце с большими внеплоскостными отклонениями.
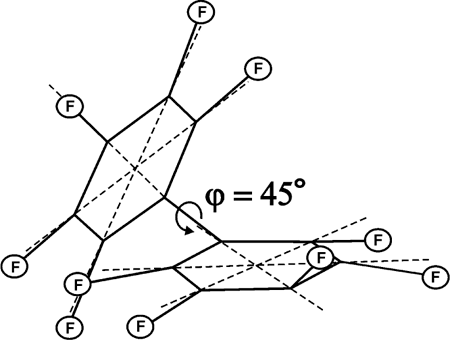
Рис. 12. Структура минимальной энергии анион-радикала перфторбифенила (C1).
Потенциальная поверхность образована двумя циклами (Рис. 13) из четырех эквивалентных минимумов (C1 структуры), разделенных переходными состояниями симметрии C2 (структуры разных циклов отличаются знаком угла φ), высота барьеров ~0.5 ккал/моль. Эти циклы связаны между собой более высокими по энергии (~4.5 ккал/моль) переходными состояниями симметрии Cs.
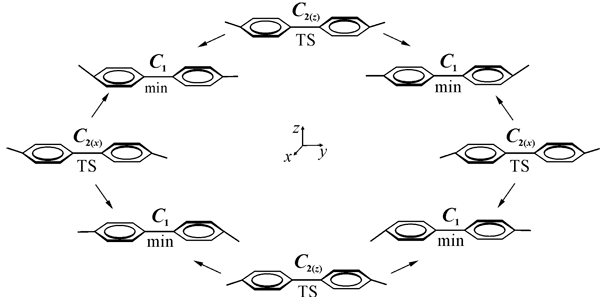
Рис. 13. Взаимосвязи низкоэнергетических структур (C6F5)2— •.
Крайне малая высота барьеров внутри каждого цикла приводит к полному усреднению СТВ в условиях ОД ЭПР экспериментов (жидкий раствор). Усреднение рассчитанных методом UB3LYP констант aF по C1 структурам приводит к величинам 71.3 (2F пара-), 18.9 (4F орто ) и 28.1 (4F мета-) Гс, которые прекрасно согласуются с экспериментальными, 72 (2F), 18.6 (4F) и 26 (4F) Гс.
Полученное экспериментальное подтверждение результатов систематических исследований потенциальных поверхностей анион-радикалов ряда полифтораренов свидетельствует об адекватности используемых моделей и указывает на необходимость учитывать сложную структуру поверхностей при интерпретации данных спектральных и химических экспериментов.
VII. Структурные искажения и механизм мономолекулярного распада анион-радикалов галогенаренов
Проведенные исследования показали, что для ион-радикалов полифторароматических молекул характерны искажения с понижением симметрии. Результатом их является качественное изменение распределения электронной плотности, способное влиять на протекание химических реакций. Энергетические барьеры, разделяющие минимумы ППЭ, по сути, являются барьерами внутримолекулярного переноса электронной плотности. Так, псевдовращение в ион-радикалах бензольного ряда представляет собой механизм переноса плотности неспаренного электрона между положениями кольца. Поэтому можно ожидать, что позиционная селективность реакций этих ион-радикалов будет в значительной мере определяться их структурной (не)жесткостью по отношению к координате псевдовращения. Характерные для анион-радикалов полифторароматических соединений внеплоскостные искажения являются механизмом переноса электронной плотности между π и σ системами. Структурные искажения этого типа могут иметь принципиальную важность для реакций, запрещенных по симметрии.
Связь между строением потенциальной поверхности и реакционной способностью исследована нами [57-61] на примере характерной реакции анион-радикалов галогенароматических соединений – фрагментации с образованием галогенид-иона – ключевой стадии синтетически важных реакций нуклеофильного замещения (SRN1) и восстановительного дегалогенирования. Для плоских π-радикалов реакция запрещена по симметрии [62] барьер ее связан с пересечением термов π и σ состояний:
ArHal — •(π-радикал) → A• (σ-радикал) + Hal—
Предполагалось [62], что переход с π- на σ-терм осуществляется за счет колебаний.
Отщепление фторид-иона протекает только в полярных растворителях, непосредственные расчеты пути реакции требуют учета сольватации. Альтернативный подход – моделирование переходного состояния в рамках газофазных расчетов – был применен к изучению распада анион-радикалов фторированных бензонитрилов [57] и бензоатов [58].
Предложенная модель переходного состояния основана на представлении об обходе пересечения π- и σ-термов за счет внеплоскостного искажения, сопровождающего растяжение связи C–F. Энергетический барьер отщепления Fˉ, Ea, в этой модели определяется положением точки π-σ-пересечения, rCFx, энергией стабилизации в этой точке за счет внеплоскостного искажения, понижением энергии переходного состояния за счет релаксации кольца и разностью энергий сольватации переходного состояния и исходного аниона (последний вклад предполагается постоянным в ряду родственных радикалов).
Выполненные в этой модели полуэмпирические INDO расчеты позволили объяснить экспериментально наблюдаемые закономерности распада анион-радикалов фторированных бензонитрилов и бензоатов, включая резкое (на шесть порядков) возрастание константы скорости реакции с накоплением атомов фтора. В обоих случаях расчеты привели к удовлетворительной линейной зависимости между барьерами реакции Ea, оцененными с помощью модельных переходных состояний, и экспериментальными величинами логарифмов констант скорости распада kd (результаты для полного ряда анион-радикалов фторбензоатов показаны на Рис. 14, специфическую сольватацию в водных растворах моделировали расчетами соответствующих бензойных кислот).
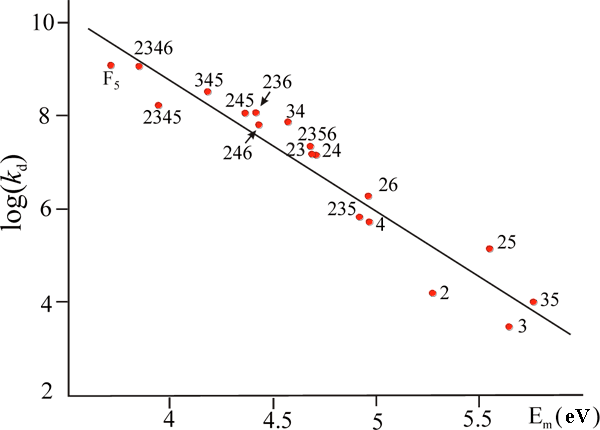
Рис. 14. Зависимость между барьерами распада анион-радикалов фторсодержащих бензойных кислот Еm (UHF/INDO) и константами скорости фрагментации kd соответствующих АР фторбензоатов, R = 0.96 (цифрами обозначены положения атомов фтора.
В рамках этой же идеологии в качестве индекса реакционной способности в [57, 58] предложено использовать величину энергетического расщепления уровней основного π- и возбужденного σ-состояний плоской структуры (ΔEπσ), ответственную за внеплоскостное искажение исходного анион-радикала. Было показано, что величины ΔEπσ могут применяться как для качественного анализа относительной способности анионов к отщеплению фторид-иона, так и для объяснения региоселективности процесса.
В рамках этого индексного подхода удалось интерпретировать данные по региоселективности гидродефторирования полифторксилолов под действием металлов (Рис. 15) на основе представлений об образовании промежуточных анион-радикалов и их последующей фрагментации.
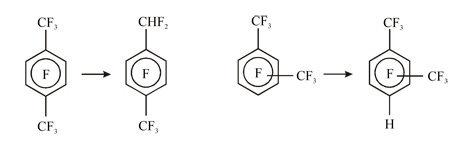
Рис. 15. Схема основных превращений изомерных перфторксилолов под действием Zn(Cu)-DMF-H2O.
Согласно результатам INDO расчетов [59] анион-радикалы перфтор-орто- и мета-ксилолов имеют более низкие энергии π→σ* возбуждения в сравнении с пара-изомером. Кроме того, положения преимущественной локализации плотности π и σ* ОЗМО аниона перфтор-пара-ксилола не совпадают (т.е. плотность электронного перехода мала). В результате, в случае анионов перфтор-мета- и орто-ксилолов, для которых ОЗМО как π, так и σ* состояний локализованы преимущественно на связях С–F ароматического кольца, происходит диссоциация этих связей при сохранении связей С–F в бензильном положении. Для анион-радикала перфтор-пара-ксилола, напротив, наиболее вероятна диссоциация связи С–F в бензильном положении.
Результаты полуэмпирических исследований [57,58] указывают на существенно неплоскую структуру переходных состояний распада анион-радикалов фторароматических соединений. Подобных структурных искажений, связанных с обходом π-σ пересечения, можно ожидать и для π-анион-радикалов других галогенароматических молекул. Для фторсодержащих анионов локализовать истинное переходное состояние распада невозможно без явного учета растворителя, так как в газовой фазе σ-терм не является диссоциативным. В то же время отщепление хлорид-аниона наблюдается и в газовой фазе [63] Удобной моделью для теоретического исследования механизма гетеролитического разрыва связи C–Hal в галогенароматических анион-радикалах представляется анион C6H5Cl— • [64].
Согласно результатам ROHF/6-31+G* исследования потенциальной поверхности C6H5Cl— •, механизм распада зависит от того, какое из анионных состояний реализуется при захвате электрона. Так, в 2A1 σ-состоянии отщепление хлорид-аниона разрешено по симметрии и осуществляется без барьера. При захвате электрона на 2B1 π-терм – отщепление Cl— из положения с максимальной плотностью неспаренного электрона – координата реакции включает внеплоскостное отклонение связи C-Cl (обход π-σ пересечения). В случае C6H5Cl— • в результате сильного вибронного взаимодействия обход осуществляется без барьера. При наиболее вероятном захвате электрона на 2A2 терм распад имеет двойной запрет по симметрии. Дополнительный запрет связан с невозможностью перехода на диссоциативный σ–терм из состояния с нулевой плотностью неспаренного электрона на связи C-Cl. Координата реакции включает плоскостное искажение кольца (обход пересечения π-термов по координате псевдовращения), при этом осуществляется перенос плотности дополнительного электрона на атом C, связанный с Cl.
Впервые локализованное расчетами переходное состояние отщепления хлорид-иона имеет неплоскую структуру и лишено всех элементов симметрии, присущих исходной молекуле C6H5Cl (C2v),60 что обуславливается сочетанием плоскостного искажения кольца и внеплоскостного отклонения разрывающейся связи. Барьер реакции связан с обходом пересечения π-термов по координате псевдовращения. Пренебрежимо малая высота барьера (0.25 ккал/моль на MP2/6-31+G*//ROHF/6-31+G* уровне расчетов) является следствием структурной нежесткости анион-радикала относительно координаты псевдовращения и согласуется с представлениями о синхронности стадий переноса электрона и разрыва связи в C6H5Cl— • [64].
Описанные в60 пути фрагментации модельного аниона C6H5Cl— • отражают возможные механизмы диссоциативного распада анион-радикалов галогенароматических соединений. Как было отмечено выше, получить истинное переходное состояние отщепления фторид-иона без явного учета сольватации невозможно. Из соображений симметрии, однако, можно утверждать, что распад C6H5F— • (основное электронное состояние 2А2) в полярных средах может реализовываться только в рамках выявленного механизма, включающего перенос электронной плотности за счет псевдовращения и внеплоскостное отклонение разрывающейся связи. Можно полагать, что и в случае C6H5F— • основным фактором, определяющим протекание реакции, является перенос электрона на разрываемую связь C–F. Сделанный прогноз подтвердился в работе Пиерини и Вера [65] при исследовании профилей энергии диссоциации связей С–Hal методом UB3LYP с учетом влияния растворителя в модели CPCM: в случае C6H5F— • переход на σ*-терм происходит не из основного, а из возбужденного π*-состояния, ОЗМО которого не имеет узловой плоскости на связи C–F. По данным [65] распад АР из 2B1-состояния происходит практически безбарьерно, таким образом, барьер мономолекулярной фрагментации есть барьер перехода с одного π-терма на другой, в наших терминах – барьер псевдовращения.
Зависимость механизма гетеролитического разрыва связи C-Hal в анион-радикалах π-типа от плотности дополнительного электрона на разрываемой связи исследована нами [61] на примере анион-радикалов изомерных хлорбензонитрилов Cl-C6H4CN на уровне расчетов ROHF, RMP2//ROHF и B3LYP в базисе 6-31+G*. Присутствие акцепторной CN-группы, стабилизирует анионы по отношению к отщеплению хлорид-иона, а также фиксирует ОЗМО b1-типа с локализацией пара- > орто- > мета- вне зависимости от положения слабо возмущающего π-систему атома Cl (Рис. 16).
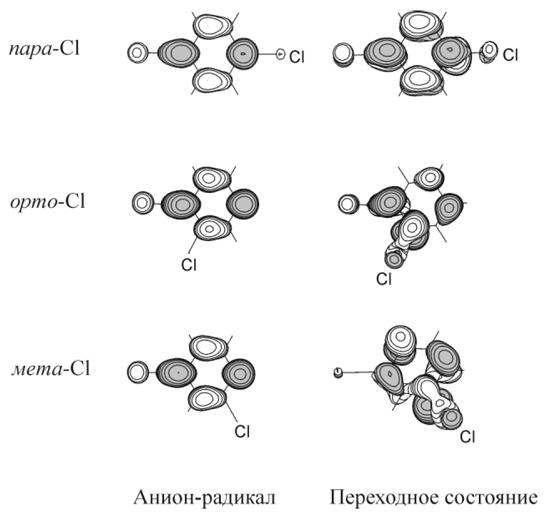
Рис. 16. Вид ОЗМО анион-радикалов Cl-C6H4CN— • и переходных состояний их распада с отщеплением аниона Cl—.
Переходные состояния распада всех трех изомерных анионов характеризуются существенными внеплоскостными отклонениями связи C-Cl. Максимальное отклонение наблюдается для мета-, а минимальное – для пара-изомера. Для пара-изомера ситуация соответствует распаду модельного аниона C6H5Cl— • при захвате электрона на 2В1 терм: координата реакции представляет собой суперпозицию растяжения связи C–Hal и ее внеплоскостного отклонения. Переходное состояние имеет симметрию Cs(yz).
В исходных структурах анион-радикалов орто- и мета-изомеров плотность ОЗМО так же локализована в пара- и ипсо-положениях относительно группы CN, в то время как в переходных состояниях распада она значительно смещена на связь C–Cl (Рис. 16). Механизмом переноса электронной плотности между положениями кольца служит движение вдоль координаты псевдовращения. Порядок изменения рассчитанных реакционных барьеров не зависит от уровня расчета и соответствует порядку изменения экспериментальных констант скорости распада анион-радикалов в жидком аммиаке [66]: пара- > орто- > мета-. Изменение относительной способности пара- и орто- изомеров к отщеплению хлорид-аниона, наблюдающееся при переходе к более полярному растворителю (H2O+tBuOH) [67], по-видимому, объясняется разной степенью локализации отрицательного заряда на уходящем атоме хлора в найденных переходных состояниях распада.
Рассмотренные эффекты обхода пересечений термов и внутримолекулярного переноса электронной плотности за счет структурных искажений, по-видимому, играют важную роль в механизмах и других запрещенных по симметрии реакций. Возникающие в ходе таких реакций структурные искажения определяются симметрией и потому существуют как в газовой фазе, так и в растворе, и не зависят от метода расчета. Заметим, что анализ пересечений термов, возможных в ходе реакции, весьма полезен с точки зрения предсказания структуры переходного состояния.
Список литературы
Материал рекомендован к публикации членом редколлегии проф. В.Е. Платоновым
Fluorine Notes, 2013, 87, 1-2


