Поступило в редакцию: апрель 2011
УДК апрель 2011
Fluorine Notes, 2012, 81, 1-2
РАДИКАЛЬНЫЕ РЕАКЦИИ ПОЛИФТОРАРЕНОВ
Л.С. Кобрина
Учреждение Российской академии наук Новосибирский институт органической химии им. Н.Н.Ворожцова
Сибирского отделения РАН, 630090, Новосибирск, пр. Лаврентьева, 9.
e-mail: kobrina@nioch.nsc.ru
Аннотация. Рассмотрены реакции полифторароматических соединений с широким кругом радикалов разной полярности и стерических требований. К этим реакциям приложимы традиционные представления, предусматривающие промежуточное образование циклогексадиенильных радикалов (полифторированных радикальных σ-комплексов). Специфика радикальных реакций полифтораренов проявляется как в преимущественной димеризации радикальных σ-комплексов, так и во впервые обнаруженной на примере этих радикалов изомеризации, реализующейся путем 1,2-миграции атома фтора.
Ключевые слова: радикальные реакции, полифторированные циклогексадиенильные радикалы, 1,2-миграция атома фтора.
Радикальные реакции являются одним из основных типов взаимодействия в органической химии, интерес к которым обусловлен тем, что свободнорадикальный механизм может реализоваться для самых разнообразных реакций, имеющих приложение в синтезе и практике.
Полифторароматические соединения с позиций изучения механизма радикальных реакций в ароматическом ряду являются принципиально новым типом объектов, позволяющих исследовать влияние на механизм радикальных реакций некоторых эффектов, отсутствующих в углеводородных системах. В первую очередь это связано с возможностью радикальной атаки по положению ароматического кольца, занятому атомом фтора, и, соответственно, с влиянием атома фтора как ипсо-заместителя на ее скорость и ориентацию, а как геминального заместителя в промежуточно образующихся радикальных σ-комплексах - на пути их превращения. Во-вторых, это вопрос совокупного влияния на указанные характеристики реакций атомов фтора, находящихся не в атакуемом, а в иных положениях ароматического субстрата и, соответственно, в ненасыщенной части интермедиата.
Систематическое исследование радикальных реакций базовых полифтораренов – гексафторбензола, октафтортолуола, декафтордифенила и октафторнафталина (ОФН) с широким кругом радикалов с варьируемыми полярностью и стерическими требованиями позволило судить о характере влияния этих факторов на основные направления радикальных реакций в ряду полифтораренов и обосновать возможность перенесения общепринятых представлений о механизме реакций этого типа в ряду нефторированных аренов и галогенаренов на полифторарены [1,2].
Реакции полифторированных соединений бензольного ряда
Для радикальных σ-комплексов, образующихся в реакциях полифторароматических соединений с алкильными и арильными радикалами, как содержащими, так и не содержащими атомов фтора, характерны три основных пути стабилизации в зависимости от полярных свойств радикала, метода его генерирования и условий проведения реакции: дефторирование с образованием продуктов гомолитического замещения, димеризация и рекомбинация.
Взаимодействие гексафторбензола в инертной атмосфере с метильным радикалом, генерированным термолизом пероксида ди-трет-бутила [3] (140°С), приводит к преимущественному образованию смесей пространственных изомеров бис(метил)-перфтортетрагидробифенилов (3а-5а), дефторированием которых с количественным выходом получены соответственно 4,4'-, 3,4'- и 3,3'-бис(метил)октафторбифенилы (6а-8а). Строение этих продуктов означает, что вслед за присоединением метильного радикала к гексафторбензолу имеет место димеризация как первоначально образующегося σ-комплекса (1а), так и образующегося из него в результате перегруппировки с миграцией атома фтора σ-комплекса (2а), а также их рекомбинация (схема 1) [3].
При взаимодействии гексафторбензола с метильным радикалом, генерированным из пероксида ацетила при 80 °С основным продуктом реакции [4,5] является смесь пространственных изомеров производного тетрагидродифенила 3а. Продукты рекомбинации 4а и 5а, обязанные своим происхождением изомеризации первоначально образующегося σ-комплекса 1а, получаются в этом случае в незначительном количестве, что обусловлено, по-видимому, легкостью разложения пероксида ацетила и, как следствие, относительно низкой температурой проведения его реакции с гексафторбензолом. Это обстоятельство приводит и к низкой скорости перегруппировки σ-комплекса с миграцией атома фтора. Другой причиной такого протекания реакции может быть более высокая скорость разложения перекиси ацетила по сравнению с перекисью ди-трет-бутила, приводящей к высокой концентрации σ-комплекса в растворе и, как следствие, понижение времени его жизни и, соответственно, понижению скорости перегруппировки. Различие путей превращения σ-комплекса, отвечающего присоединению к гексафторбензолу метильного радикала, генерированного из разных источников (ароматизация [6] в реакциях С6F6 с СН3NO2 при 550°С и рекомбинация [3] в реакциях с пероксидами ацетила и ди-трет-бутила), определяется наличием или отсутствием в реакционной среде радикала, способного эффективно отрывать атом фтора от sp3-гибридного атома углерода радикального σ-комплекса.
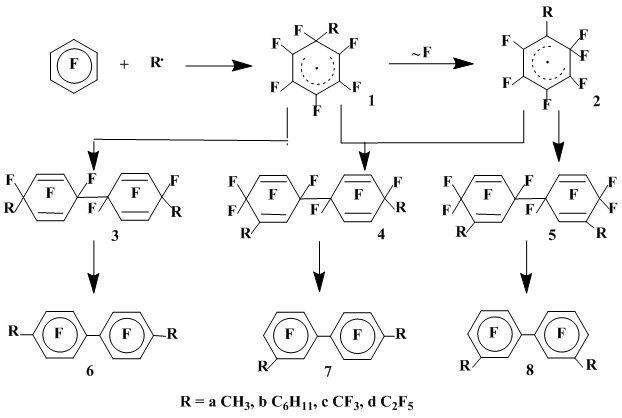
Схема 1
Гомолитическое замещение атома фтора в полифторароматических соединениях становится основным направлением реакции, когда в качестве источника метильного радикала используется система метилмагнийиодид - галогенид серебра или меди в диэтиловом эфире (механизм генерирования метильного радикала в этих условиях описан в работах [7,8,9]), что связано с наличием в реакционной среде агентов, эффективных в отношении ароматизации радикальных σ-комплексов с удалением геминального атома фтора, например, восстановленных металлов. Наряду с метилсодержащими соединениями в этой реакции образуются продукты замещения атома фтора в полифтораренах вторичным радикалом - этоксиэтильным, который генерируется, по-видимому, в результате атаки метильным радикалом диэтилового эфира [10,11]:
СН3МgI + МХ → СН3М + MgIX
СН3М → M +
.CH3
.CH3 + CH3CH2OCH2CH3 → CH4 + CH3.CHOCH2CH3
Изомерный состав продуктов метилирования октафтортолуола характеризуется соотношением о:м:п = 37:24:39. В ряду субстратов типа С6F5Х (X = F, СF3, С6F5) наблюдается зависимость соотношения двух указанных конкурирующих реакций радикального замещения фтора [12] от характера заместителя X: при переходе от X = F к X = СF3 и далее к X = С6F5 уменьшается выход метилзамещенных и увеличивается выход 1-этоксиэтилзамещенных продуктов, причем в случае декафторбифенила образуются почти исключительно изомерные 1-этоксиэтилнонафторбифенилы (схема 2).
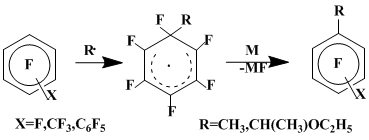
Схема 2
Гексафторбензол взаимодействует с циклогексильным радикалом, генерированным термическим разложением (140°С) ди-трет-бутилпероксида в циклогексане, преимущественно по пути, включающем рекомбинацию и димеризацию первично образующихся σ-комплексов 1б и их изомеров 2б, возникающих в результате миграции атома фтора из sp3-гибридного узла в соседнее положение (схема 1). Подобно приведенным выше случаям реакций гексафторбензола с метильным радикалом, генерированным термолизом пероксидов, продукты рекомбинации σ-комплексов присоединения к С6F6 циклогексильного радикала являются смесями пространственных изомеров [13], дефторирование которых приводит к изомерным дициклогексил-октафторбифенилам 6б - 8б.
Однако проведение реакции гексафторбензола с циклогексильным радикалом в присутствии соединений, обеспечивающих возможность ароматизации первичного σ-комплекса (димера пентафторфеноксильного радикала, молекулярного кислорода, гидропероксида ди-трет-бутила), приводит к образованию наряду с продуктами рекомбинации σ-комплексов 3б-5б заметных количеств циклогексилпентафторбензола (20%). Агентом, отрывающим атом фтора от радикального σ-комплекса, является, вероятно, пентафторфеноксильный радикал, превращающийся при этом в гексафторциклогексадиенон. Возможность участия пентафторфеноксильного радикала в ароматизации радикальных σ-комплексов, связанной с отрывом атома фтора, находится в согласии с данными об эффективности пентафторфенола в качестве добавки, увеличивающей вклад процесса замещения при фенилировании полифторароматических соединений [14]. В условиях радикальной реакции пентафторфенол может легко превращаться в пентафторфеноксильный радикал [15] (схема 3).
В процесс ароматизации могут легко вовлекаться как первичные σ-комплексы, так и их изомеры, образующиеся в результате перегруппировки с миграцией атома фтора. При этом соотношение продуктов димеризации и рекомбинации σ-комплексов не зависит от того, проводится реакция в атмосфере инертного газа или в присутствии кислорода воздуха. Такое, очевидно, возможно лишь в том случае, если оба типа σ-комплексов дефторируются примерно с одинаковой скоростью.
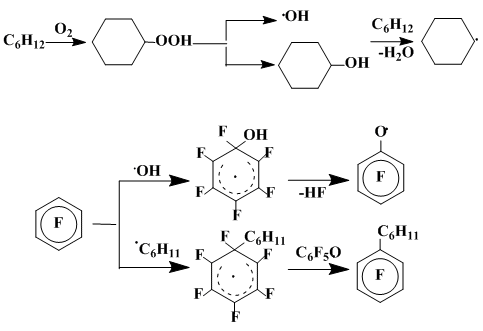
Схема 3
При взаимодействии гексафторбензола с трифторметильным и перфторэтильным радикалами, генерированными термолизом (80°С) соответственно пероксидов перфторацетила и перфторпропионила [3,4,5], в качестве основных продуктов реакции образуются димеры σ-комплексов, являющиеся, по-видимому, смесями пространственных изомеров. Их дефторирование с количественным выходом приводит к перфтор-4-4'-бис(алкил)бифенилам 6c,d (схема 1).
Строение полученных полифторбифенилов свидетельствует о том, что, в отличие от аналогичных реакций водородсодержащих алкильных радикалов, σ-комплексы, образующиеся при присоединении к гексафторбензолу перфторалкильных радикалов, не претерпевают перегруппировки с миграцией атома фтора.
Довольно сложно протекает взаимодействие декафторбифенила с трифторметильным радикалом, генерированным термолизом пероксида трифторацетила [16].
Наряду со сложной смесью димеров σ-комплексов (13%), отвечающих присоединению трифторметильного радикала к декафторбифенилу, дефторированием которой количественно получены перфторбис(фенил)бис(метил)бифенилы, образуется смесь перфторфенил(диметил)циклогексадиенов (9-11) (18%), являющихся смесями геометрических изомеров, обусловленных взаимным пространственным расположением трифторметильных групп. Образование указанных соединений означает, что радикальные σ-комплексы, образующиеся в ходе этой реакции, стабилизируются не только путем димеризации, но и путем рекомбинации с трифторметильными радикалами (схема 4).
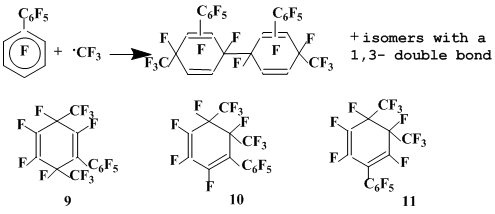
Схема 4
Как и в случае реакций гексафторбензола с .СF3 и .С2F5, σ-комплексы, образующиеся в результате присоединения .СF3 к декафторбифенилу, не претерпевают перегруппировки с 1,2-миграцией атома фтора.
Взаимодействие гексафторбензола с перекисью бензоила в зависимости от условий реакции [17] приводит к образованию от 6 до 50% димеров радикальных σ-комплксов присоединнения фенильного радикала к гексафторбенззолу (12 и 13), дефторирование которых дает кватерфенилы (16 и 17), строение которых свидетельствует о перегруппировке первоначального σ-комплекса с 1,2-миграцией атома фтора (таблица 1, схема 5).
Таблица 1. Взаимодействие перекиси бензоила с гексафторбензолом [17]
-
Концентрация перекиси
Т °С
Время, час
Выход %
Соотношение
16 и 17
C6H5C6F5
C24H10F12
0.0125
0.025
0.025
60
80
80
200
72
100
17
43.5
62.5
50
25
6
49:39
22:42
8:55
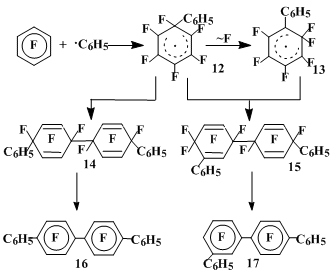
Схема 5
При взаимодействии гексафторбензола с пероксидом пентафторбензоила практически единственным направлением реакции является образование продуктов рекомбинации и димеризации радикальных σ-комплексов. При 80°С в основном (выход 70%) образуется димер (21) σ-комплекса (18), отвечающего присоединению к гексафторбензолу пентафторбензоилоксильного радикала [18], наряду с которым выделены декафторбифенил и перфторфенилбензоат (в сумме 5%) и незначительное количество смеси продуктов рекомбинации σ-комплексов (22) и (23), содержащих как пентафторбензоилоксильную, так и пентафторфенильную группу (схема 6).
Повышение температуры проведения реакции (80-200°С) приводит к образованию соединений, отвечающих атаке субстрата пентафторфенильным радикалом [19,20]. При 200°С основным продуктом [19] является димер (26) σ-комплекса (20). Строение димера 26, доказанное спектрально и подтвержденное количественным дефторированием до м,м'-перфторкватерфенила (29), свидетельствует о том, что первоначально образующийся в результате присоединения пентафторфенильного радикала к гексафторбензолу σ-комплекс (19) претерпевает перегруппировку с миграцией атома фтора из геминального узла в соседнее положение. Этот результат — первое свидетельство 1,2-сдвига атома фтора от sр3-гибридиого атома углерода к соседнему радикальному центру, что является наиболее важной отличительной чертой радикальных реакций полифторированных ароматических соединений.
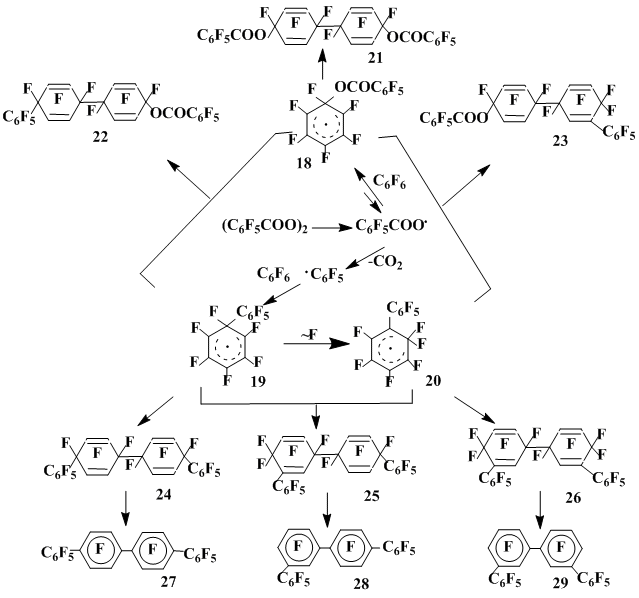
Схема 6
В интервале температур 80-200°С (таблица 2) можно получить все возможные продукты 21-26 димеризации и рекомбинации трех радикальных σ-комплексов 18-20, генерирующихся в ходе взаимодействия гексафторбензола с пероксидом пентафторбензоила [20] (в отличие от этого, авторам [21,22] удалось идентифицировать в качестве продуктов этой реакции только декафторбифенил и перфторфенилбензоат с общим выходом 10%. Из представленных в таблице 2 зависимостей видно, что при повышении температуры и уменьшении концентрации пероксида увеличивается доля продуктов перегруппировки 23, 25 и 26 и уменьшается доля димера 21. Увеличение степени перегруппировки с уменьшением концентрации пероксида при фиксированной температуре можно связать с увеличением времени жизни радикальных σ-комплексов в результате уменьшения их концентрации в растворе.
При высоких концентрациях пероксида уменьшается время жизни неперегруппированных σ-комплексов, что приводит к резкому уменьшению концентрации перегруппированных σ-комплексов (см. в таблице 2 содержание димеров 23 и 26).
Таблица 2. Разложение пероксида пентафторбензоила в гексафторбензоле [20]
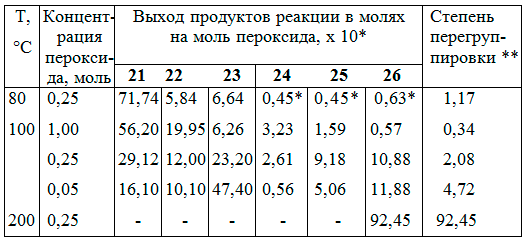
* Выходы определены по данным ГЖХ продуктов дефторирования смеси димеров 24, 25, 26
** Рассчитана по соотношению: 26 + 1/2 25 : 24 + 1/2 25
Повышение температуры и уменьшение концентрации пероксида сказывается на соотношении выходов продуктов превращений σ-комплексов, отвечающих присоединению пентафторфенильного и пентафторбензоилокснльного радикалов к гексафторбензолу таким образом, что увеличивается содержание первых. Это может быть связано с обратимостью присоединения к гексафторбензолу пентафтор-бензоилоксильного радикала и возрастанием скорости его декарбоксилирования. Изучение термолиза димера 21 в гексафторбензоле и бензоле [20] при 200°С дает четкие свидетельства обратимости присоединения пентафторбензоилоксильного радикала к гексафторбензолу.
Вопрос об обратимости или необратимости присоединения пентафторфенильного радикала к ароматическому кольцу является одним из центральных при обсуждении механизма его взаимодействия с полифторароматическими соединениями. Описывающая процесс разложения пероксида пентафторбензоила в гексафторбензоле схема основана на представлениях о необратимости этого процесса, а также необратимости димеризации и рекомбинации всех образующихся при этом радикальных σ-комплексов в условиях реакции (80, 100°С). Последнее обосновано тем, что соединения 24-26 остаются неизменными [20] после длительной выдержки (45ч) в абсолютном бензоле при 100°С.
Кроме того, показано [20], что при неполном термолизе димера 24 при 200°С в продуктах реакции присутствует только исходный димер и димер 26 (по данным ГЖХ продуктов термолиза после их дефторирования). Отсутствие соединения 25 среди продуктов термолиза означает, что σ-комплекс 19 в этих условиях быстро перегруппировывается в σ-комплекс 20. Эти результаты показывают, что присоединение пентафторфенильного радикала к гексафторбензолу практически необратимо вплоть до 200°С. При 220°С за 20 ч димер 24 полностью превращается в димер 26. Гексафторбензол среди продуктов реакции не обнаружен.
Димер 26 не изменяется за 20 ч при 200°С, что означает необратимость в этих условиях суммарного процесса, включающего последовательно присоединение пентафторфенильного радикала к гексафторбензолу и перегруппировку образующегося при этом радикального σ-комплекса и димеризацию продукта перегруппировки. Однако при 250°С соединение 26 превращается в декафторбифенил [20].
Использование других потенциальных источников пентафторфенильного радикала с целью осуществления его взаимодействий с гексафторбензолом не дало столь информативных результатов. Так, не реагирует с С6F6 .С6F5, генерированный фотолизом пентафториодбензола [23] или окислением пентафторфенилгидразина оксидом серебра [24] при 0°С. Однако при 100°С последняя реакция приводит к образованию упомянутых выше продуктов взаимодействия гексафторбензола с пентафторфенильным радикалом 24-26, хотя их выход и не велик, а основным направлением реакции является атака пентафторфенильным радикалом молекулы исходного пентафторфенилгидразина, приводящая к образованию пентафторбензола [25].
Кроме этого из реакционной смеси был выделен перфтор-4,4'-бис(фенил)-1,1'-бициклогексадиенилиден (32) в виде смеси двух пространственных изомеров. Дефторирование этого соединения цинковой пылью в ледяной уксусной кислоте приводит с высоким выходом к п,п'-перфторкватерфенилу (27). Образование соединения 32 связано, по-видимому, с последовательной реализацией присоединения .С6F5 к пентафторфенилгидразину, окисления образующегося при этом σ-комплекса (30) оксидом серебра, приводящего в конечном счете к диенокарбену (31), димером которого является соединение 32. Аналогией такой схемы может служить генерирование кетокарбена из хинондиазидов [26] (схема 7).
При использовании пентафторфенилазотрифенилметана в качестве источника пентафторфенильного радикала (80°С) в реакции с гексафторбензолом получена сложная смесь продуктов, содержащая следы декафторбифенила и димера 26. По-видимому, образующийся в ходе процесса пентафторфенильный радикал рекомбинирует с трифенилметильным радикалом, стационарная концентрация которого достаточно высока, или взаимодействует с нефторированной частью молекулы пентафторфенил-азотрифенилметана [3].
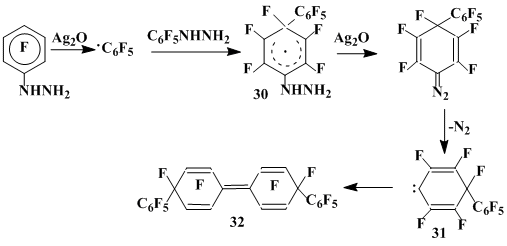
Схема 7
Изучение реакций октафтортолуола и декафторбифенила с пероксидом пентафторбензоила дало возможность получить картину, характеризующую влияние электронных и стерических эффектов трифторметильной и пентафторфенильной групп на закономерности радикальных реакций, выявленные на примере гексафторбензола, в частности, на относительную способность радикальных σ-комплексов к димеризации и перегруппировке с миграцией атома фтора. Взаимодействие октафтортолуола [27] и декафторбифенила [28] с пероксидом пентафторбензоила, в отличие от аналогичной реакции гексафторбензола, даже в мягких условиях (80°С) приводит к образованию с высокими выходами продуктов изомеризации и рекомбинации σ-комплексов, отвечающих присоединению пентафторфенильного радикала преимущественно в мета-положение по отношению к отличному от фтора заместителю, наряду с небольшими количествами продуктов, характеризующегося такой же региоселективностью гомолитического замещения: перфторметилбифенилов в реакции октафтортолуола и перфтортерфенилов в реакции декафторбифенила (схема 8).
При дефторировании димеров, полученных в реакциях октафтортолуола, наряду с перфторбис(фенил)бис(метил)бифенилами образуются перфторметилбифенилы, что может указывать на частичную диссоциацию димеров с образованием радикальных σ-комплексов (схема 9).
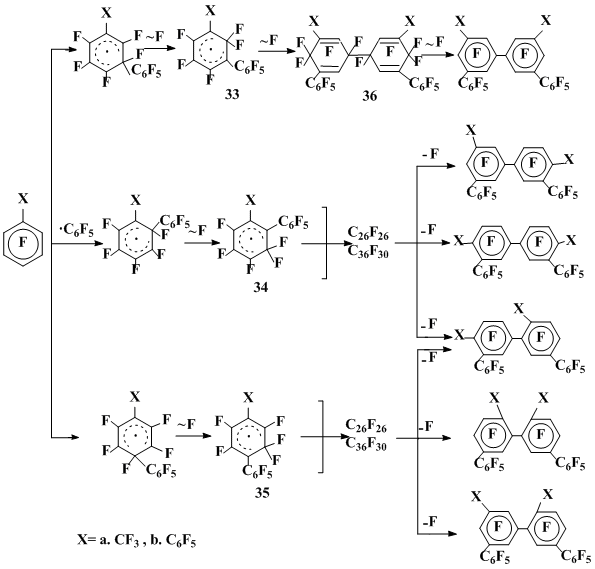
Схема 8
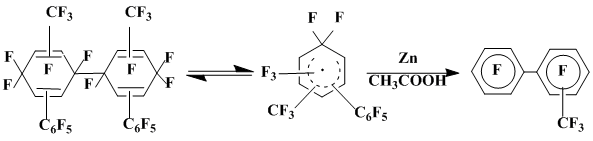
Схема 9
Возможность такой диссоциации подтверждается тем, что в масс-спектрах димеров радикальных σ-комплексов линия, отвечающая молекулярному иону, имеет малую интенсивность или вообще отсутствует, а наиболее интенсивной является линия, соответствующая иону с м/е 1/2М.
Наряду с изомерными продуктами рекомбинации σ-комплексов C26F26 взаимодействие пероксида пентафторбензоила с октафтортолуолом [27] приводит также к образованию заметного количества смеси соединений состава C45F44. Судя по структуре (38), приписанной выделенному из этой смеси индивидуальному соединению по результатам его дефторирования (соединение 39), они являются продуктами взаимодействия димера (36а) и изомерных ему соединений с пентафторфенильным радикалом и последующей рекомбинации образующихся радикальных σ-комплексов типа (37) с σ-комплексом (33а) (схема 10).
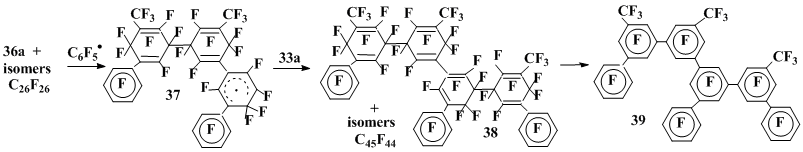
Схема 10
Несколько неожиданным кажется то, что взаимодействие пентафторфенильного радикала с димерами C26F26 идет не по двойной связи, а по ароматическому кольцу, поскольку известно, что полифторированные олефины реагируют с углеводородными радикалами с большей скоростью, чем полифторированные ароматические соединения [29]. Отсутствие реакции по двойной связи может быть связано как со стерическими препятствиями радикальной атаке, так и с меньшей возможностью для делокализации плотности неспаренного электрона в отвечающем этому направлению реакции интермедиате, чем при атаке по ароматическому кольцу.
Как и следовало ожидать, в жестких условиях (200°С) из-за уменьшения субстратной селективности радикальной атаки такой побочный процесс протекает в меньшей степени. Повышение температуры проведения реакции пероксида пентафторбензоила с октафтортолуолом сказывается также на выходе и изомерном составе образующихся перфторметилбифенилов. Увеличение выхода этих продуктов при повышении температуры, по-видимому, свидетельствует о большем ускорении процесса ароматизации радикальных σ-комплексов по сравнению с иными их превращениями, а изомерный состав продуктов замещения фтора, реализующийся при 200 °С (примерно равные количества о-, м- и п-перфторметилбифенилов), отражает уменьшение селективности атаки субстрата пентафторфенильным радикалом с ужесточением условий реакции. Данные, полученные ранее [30] для реакций с фенильным радикалом нефторированных ароматических соединений, позволяют считать, что в этих условиях миграции пентафторфенильного кольца не происходит.
Отсутствие в реакциях октафтортолуола и декафторбифенила с пероксидом пентафторбензоила продуктов, обязанных своим образованием атаке субстрата пентафторбензоилоксильным радикалом, является, по-видимому, следствием меньшей реакционной способности этих субстратов по сравнению с гексафторбензолом по отношению к электрофильному пентафторбензоилоксильному радикалу. Кроме того, определенную роль, очевидно, может играть меньшая стабильность продуктов рекомбинации σ-комплексов, отвечающих присоединению пентафторбензоилоксильного радикала к октафтортолуолу и декафторбифенилу, а также предшествующих им σ-комплексов, по сравнению с их аналогами, образующимися из гексафторбензола, в отношении обратимости их образования. В сочетании этого с практической необратимостью декарбоксилирования пентафторбензоилоксильного радикала и возможен наблюдаемый результат.
Выявленная для реакций октафтортолуола и декафторбифенила с пероксидом пентафторбензоила региоселективность позволяет предположить, что относительная стабильность изомерных σ-комплексов, отвечающих присоединению пентафтор-фенильного радикала к этим субстратам, соответствует большей предпочтительности участия атома фтора в делокализации плотности неспаренного электрона по сравнению с трифторметильной и пентафторфенильной группами. Следствием этого, очевидно, является большая эффективность стабилизирующего влияния атомов фтора в резонансных положениях ненасыщенной части полифторированных циклогексадиенильных радикалов, чем влияние указанных заместителей. В случае пентафторфенильной группы это может быть связано с некопланарностью перфторированных фенильного и циклогексадиенильного фрагментов.
Рассмотренная выше качественная картина относительной реакционной способности гексафторбензола, октафтортолуола и декафторбифенила, выявленная по результатам реакций этих соединений с пентафторфенильньм радикалом, находит подтверждение при оценке реакционной способности ароматических соединений в радикальных реакциях с использованием подхода, развитого в [31]. Хорошее согласие расчетных величин относительной реакционной способности гексафторбензола и бензола в радикальных реакциях с данными эксперимента, а также хорошо просматриваемая тенденция зависимости реакционной способности субстрата от полярных свойств радикалов показывают [32,33] плодотворность применения такого подхода для характеристики радикальных реакций полифтораренов (в ряду радикалов .СН3, .С6Н5, .СF3, .С6F5 возрастает электрофильность и в этом же ряду падает относительная скорость присоединения этих радикалов к С6F6 по сравнению с С6Н6).
Взаимодействие гексафторбензола с гидроксильным радикалом, генерированным термолизом (140°С) 90%-й Н2О2 в растворе ацетонитрила [34], приводит к образованию пентафторфенола, дальнейшее окисление которого избытком пероксида водорода дает дифтормалеиновую кислоту. Если термолиз пероксида водорода осуществлять в избытке пентафторфенола, то основным продуктом реакции становится тетракис-(пентафторфенокси)бензохинон-1,4 [34] (схема 11).
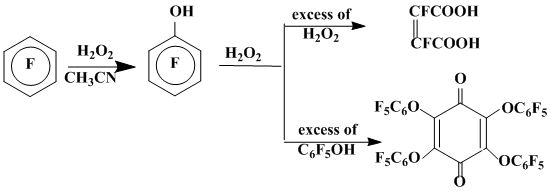
Схема 11
В аналогичных условиях из пентафторбифенила, в котором имеется возможность конкурентной атаки гидроксильным радикалом по атомам углерода, связанным как с атомами фтора, так и с атомами водорода, преимущественно образуются изомерные пентафторфенилфенолы [34], что свидетельствует о предпочтительности реакции электрофильного гидроксильного радикала по незамещенному ароматическому кольцу.
Реакции октафторнафталина
При взаимодействии октафторнафталина с различными источниками метильных радикалов происходит как гомолитическое замещение атомов фтора в положениях 1 и 2, так и димеризация радикальных σ-комплексов, образующихся в ходе реакции в результате присоединения этих радикалов к октафторнафталину. Так, термолиз пероксида ди-трет-бутила в ОФН (140°С) в атмосфере инертного газа приводит к образованию смеси продуктов рекомбинации таких σ-комплексов (40), а также 2-метилгептафторнафталина (41) (выходы соответственно 34 и 15% в расчете на вступивший в реакцию ОФН) [35]. При более высокой температуре (160°С) в дополнение к этим продуктам реакции, выходы которых увеличиваются (54 и 36% соответственно), появляется незначительное количество 1-метилгептафторнафталина (42) (5%) (схема 12).
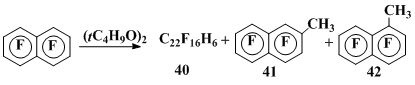
Схема 12
В присутствии кислорода воздуха взаимодействие ОФН с пероксидом ди-трет-бутила приводит к образованию исключительно продукта гомолитического замещения фтора - 2-метилгептафторнафталина, то есть основным направлением превращения радикального σ-комплекса становится дефторирование, подобно тому, что обсуждалось для реакций гексафторбензола с алкильными радикалами в присутствии кислорода воздуха.
При использовании в реакции с ОФН в качестве источника метильного радикала метилмагнийиодида в комбинации с галогенидами серебра или меди в диэтиловом эфире основным направлением, как и для описанных выше реакций соединений бензольного ряда, является гомолитическое замещение атома фтора. При этом наряду с метилгептафторнафталинами образуются также продукты замещения атома фтора вторичным радикалом — этоксиэтильным. Соотношение 1- и 2-метил-гептафторнафталинов равно 90:10, а смесь α-этоксиэтилгептафторнафталинов содержит примерно равные количества 1- и 2-изомера [12]. При степени конверсии октафторнафталина 40-55% соотношение метил- и α-этоксиэтилгептафторнафталинов при использовании CuI не зависит от мольного соотношения СН3МgI/ОФН (1:5 и 1:10) и равно 2:1, а в реакции с применением АgСl при увеличении концентрации ОФН в 2 раза становится равным 4:1.
Обращает на себя внимание тот факт, что ориентация в реакциях октафторнафталина с метильным радикалом, приводящих к замещению атома фтора, зависит от метода генерирования метильного радикала: при использовании в качестве его источника ди-трет-бутилпероксида преимущественно образуется 2-метилгептафторнафталин, а если таковым является система СН3МgI-СuI(АgСl) - 1-метилгептафторнафталин. Разница в температурах, при которых проводятся эти реакции (140°С в первом случае и 25°С - во втором), послужила основанием для предположения о том, что причиной изменения региоселективности является обратимость присоединения метильного радикала к ОФН при высокой температуре (140°С) и необратимость перегруппировки с миграцией атома фтора, имеющей место только для σ-комплекса, отвечающего присоединению .СН3 в положение 2 октафторнафталина, что сдвигает реакцию при наличии в системе дефторирующих агентов в сторону образования 2-метилгептафторнафталина. При дальнейшем повышении температуры (160°С) увеличивается скорость дефторирования и в реакционной смеси, как уже отмечалось, появляются наряду с 2-изомером небольшие количества 1-метилгептафторнафталина (схема 13).
Проведение реакции при низкой температуре в условиях необратимости присоединения метильного радикала к октафторнафталину дает кинетически контролируемую смесь изомерных метилгептафторнафталинов, в которой преобладает 1-изомер 42.
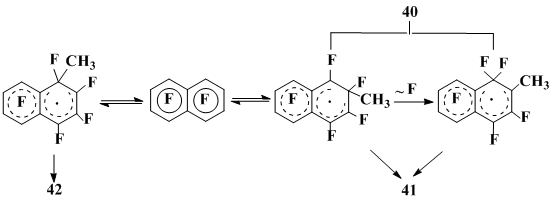
Схема 13
При взаимодействии октафторнафталина с пероксидом бензоила при 85°С образуются продукты замещения фтора - 1-и 2-фенилгептафторнафталины в соотношении 3.3:1 и 1- и 2-бензоилоксигептафторнафталины в соотношении 1:1, наряду с перфтор-2-(1'-нафтокси)нафтохиноном-1,4 (46), гексафторнафтохиноном (47) и продуктами превращения пероксида бензоила (бифенил, фенилбензоат, фторангидрид бензойной кислоты) [36].
В среде ацетонитрила [34] основной в этой реакции является смесь продуктов (45) димеризации и рекомбинации σ-комплексов (43) и (44), отвечающих присоединению к октафторнафталину как бензоилоксильного, так и фенильного радикалов (схема 14).
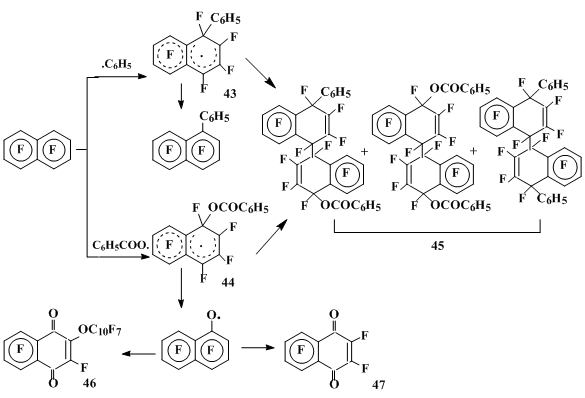
Схема 14
Реакция октафторнафталина с пентафторбензолсульфофторидом в качестве источника пентафторфенильного радикала при 160°С приводит к образованию перфтор-2-фенилнафталина (10%) (незначительные количества 1-изомера (2%) образуются только при проведении реакции при 200°С) и смеси (50) (34%) продуктов рекомбинации и димеризации как σ-комплекса (48), отвечающего присоединению пентафторфенильного радикала в положение 2 ОФН, так и σ-комплекса (49), образующегося из σ-комплекса 48 в результате перегруппировки с миграцией атома фтора [35] (схема 15).
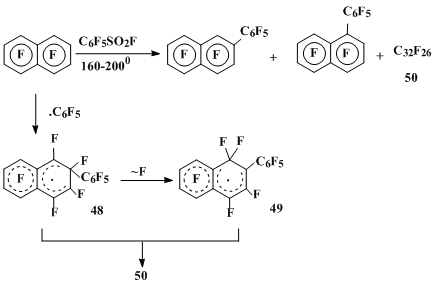
Схема 15
Взаимодействие октафторнафталина с пероксидом пентафторбензоила в растворе ацетонитрила [37] при 80°С приводит к преимущественному образованию продукта димеризации (52) σ-комплекса (51), отвечающего присоединению пентафтор-бензилоксильного радикала в положение 1 октафторнафталина. Процесс, по-видимому, обратим, поскольку при 120°С димер 52 превращается в октафторнафталин и смесь соединений того же состава, что и при проведении реакции при 120°С в отсутствие растворителя. Основными компонентами смеси продуктов взаимодействия ОФН с пероксидом пентафторбензоила при 120°С в отсутствие растворителя [38] являются перфтор1-оксо-2-(нафтокси-1')дигидронафталин-1,4 (55) и перфтор-1-оксо-2-(бензоил-окси)дигидронафталин-1,4 (56). Кроме того, образуются фторангидрид пентафтор-бензойной кислоты, гексафторнафтохинон-1,4 47 и небольшие количества (1,5%) продуктов гемолитического замещения атомов фтора - 1- и 2-перфторнафтилбензоаты. Продуктов участия в этой реакции пентафторфенильного радикала не было обнаружено.
Образование кислородсодержащих соединений в реакциях ОФН с пероксидом пентафторбензоила не имеет аналогий среди известных реакций фторированных соединений и связан, вероятно, с генерированием в условиях реакции гептафторнафтоксильного радикала. Выделение из продуктов этой реакции фторангидрида пентафторбензойной кислоты является серьезным доводом в пользу схемы, по которой гептафторнафтоксильный радикал (53) генерируется в результате элиминирования фторангидрида пентафторбензойной кислоты из σ-комплекса 51. Вероятные пути дальнейших превращений гептафторнафтоксильного радикала показаны на схеме 16.
Основными продуктами реакции октафтонафталина с пероксидом ди-трет-бутила при 110°С и высокой концентрации пероксида (0,25 М) или с пентафторбензол-сульфогалогенидами в присутствии меди и ее солей являются 1-оксопроизводные 1,2- и 1,4-полифтордигидронафталинов, также, по-видимому, обязанные своим образованием превращениям гептафторнафтоксильного радикала, который генерируется в результате фрагментации радикальных σ-комплексов, образующихся при присоединении к ОФН трет-бутоксильного или пентафторфенилсульфонильного радикала [35] (схема 17).
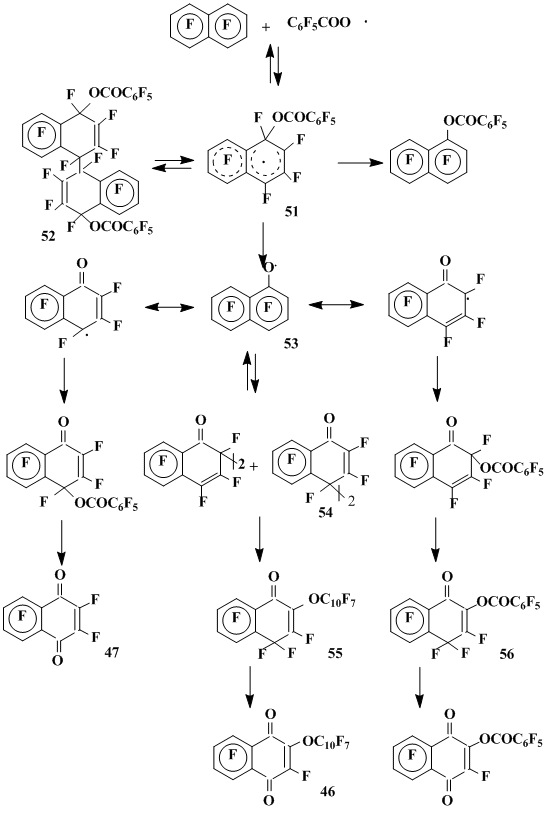
Схема 16
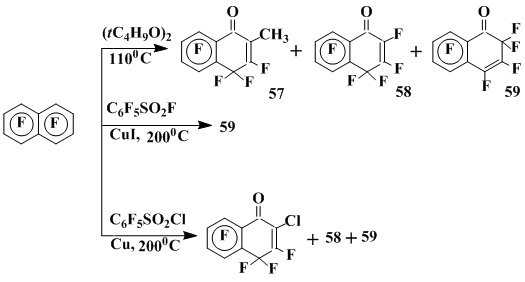
Схема 17
Состав продуктов термолиза пероксида водорода в октафторнафталине (100°С), а также в гептафторнафтолах-1 и -2 косвенно подтверждает предположение об ответственности гептафторнафтоксильного радикала за образование кислородсодержащих продуктов в реакциях октафторнафталина [39] (схема 18).
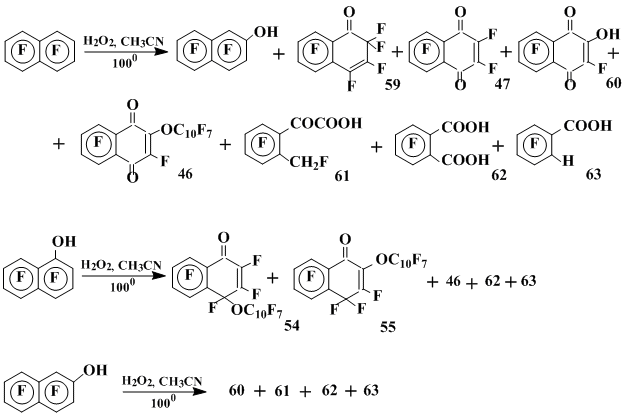
Схема 18
На схеме 18 приведены основные продукты этих реакций: гептафтор-2-нафтол, 1-оксоперфтор-1,2-дигидронафталин (59), гексафторнафтохинон-1,4 47, 2-гидрокси-пентафторнафтохинон-1,4 (60), перфтор-2-(1'-нафтокси)-1,4-нафтохинон (57) и 2,3,4,5-тетрафтор-6-(фторметил)фенилглиоксиловая кислота (61). При большом избытке перекиси водорода идет более глубокое окисление с образованием наряду с соединениями 47, 57, 59 и 60, тетрафторфталевой (62) и 2,3,4,5-тетрафторбензойной (63) кислот (схема 18).
Примеры наиболее важных превращений гептафторнафтоксильных радикалов приведены на схеме 19.
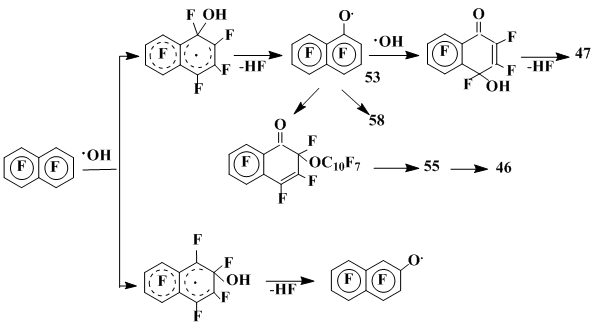
Схема 19
Пути трансформации полифторированных радикальных σ-комплексов
Для взаимодействия полифторароматических соединений со свободными радикалами характерны следующие основные типы превращений образующихся в этих реакциях радикальных σ-комплексов, характеризующихся общностью первой стадии - ипсо-присоединением радикала по положению субстрата, занимаемому атомом фтора (схема 20).
- ароматизация σ-комплекса с отщеплением радикала R и возвратом к исходному субстрату;
- ароматизация с отрывом атома фтора и образованием продукта гомолитического замещения;
- фрагментация радикального σ-комплекса с отщеплением диамагнитной молекулы из геминального узла и образованием нового полифторированного радикала;
- рекомбинация полифторированных циклогексадиеннльных радикалов, в том числе димеризация;
- неизвестная ранее в ряду фторсодержащих радикалов перегруппировка полифторированных циклогексадиенильных радикалов с 1,2-миграцией атома фтора из геминального узла к соседнему радикальному центру.
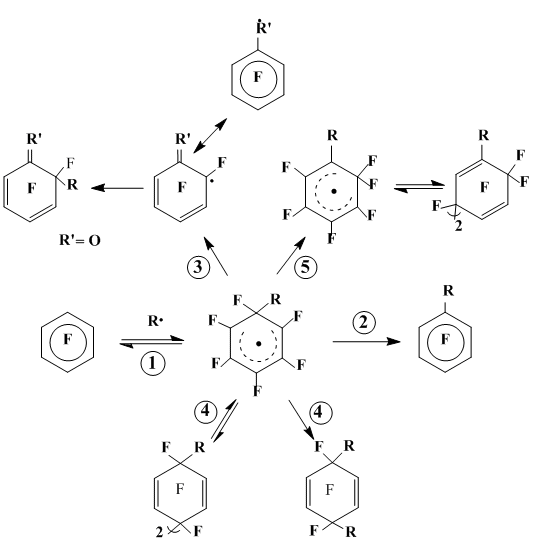
Схема 20
При рассмотрении закономерностей протекания первой стадия радикальных реакций полифторароматическнх соединений первостепенное значение имеют факторы, определяющие скорость присоединения радикалов к субстратам с образованием радикальны: σ-комплексов и возможность их возврата к исходному соединению, а также соотношение скоростей такого превращения σ-комплекса и других возможных путей его стабилизации. Со стороны атакующего радикала основную роль на этой стадии играет его полярность, а со стороны субстрата — число и взаимное расположение атомов фтора.
Различные типы конкурентных экспериментов по выяснению соотношения реакционной способности гексафторбензола и бензола в растворе приводят к результатам, подтверждающим важную роль полярных свойств атакующего радикала. Так, электрофильные радикалы присоединяются к бензолу с большей скоростью, чем к гексафторбензолу, тогда как для нуклеофильных радикалов наблюдается противоположная картина. Например, соотношение скоростей присоединения к С6F6 и С6Н6 составляет для фенильного радикала [40] 0,44 при 80°С, для метильного радикала [29] 7.8 при 65°С, для трифторметильного [29] 0,4 и для α,α-дифторэтильного [29] 0,8 при 50 °С.
Несмотря на то, что представление о ключевой роли циклогексадиенильных радикалов как интермедиатов радикальных реакций ароматических соединений сложилось достаточно давно, до недавнего времени об их образовании в таких процессах судили только по косвенным признакам - наличию в продуктах реакций соединений, структура которых отвечала димеризации σ-комплексов, а также на основании результатов, полученных с помощью метода спиновых ловушек. Детектирование и прямое наблюдение этих интермеднатов долгое время были ограничены специфическими случаями, когда вследствие стерических затруднений, обусловленных, например, наличием нескольких трет-бутильных [41] или триметилсилилоксильных групп [42], такие радикалы обладают повышенной стабильностью, а также интермедиатами реакций присоединения гидроксильного радикала к ароматическому ядру [43,44,45].
Были получены также спектры ЭПР циклогексадиенильных радикалов, образующихся в результате присоединения трифенилсилильного радикала к бензонитрилу [46], силильных, фосфонильных и метильных радикалов к бензолу [47].
Совокупность всех этих подходов применена и для доказательства образования циклогексадиенильных интермедиатов радикальных реакций полифторароматических соединений, что делает степень обоснованности представлений о механизме этих реакций не меньшей, чем для нефторированных аренов.
Метод спиновых ловушек хорошо зарекомендовал себя при изучении механизма гомолитических реакций и установлении природы образующихся в ходе этих реакций короткоживущих радикалов [48-53]. Данных, касающихся изучения реакций этого типа в ряду полифторированных соединений с использованием спиновых ловушек, немного. Однако они позволяют судить о природе радикалов, образующихся при термическом разложении фторсодержащих бензоилпероксидов или фенилазотрифенилметанов в ароматическим субстратах (бензол или гексафторбензол) в присутствии С-фенил-N-трет-бутилнитрона (ФБН) и нитрозодурола (НД) в качестве спиновых ловушек, а также соответствующих их присоединению к субстратам циклогексадиенильных радикалов.
Полученные нами параметры спектров ЭПР спиновых аддуктов фторсодержащих радикалов [54] показывают, что независимо от количества атомов фтора в ароилоксильных радикалах, образующихся при разложении фторзамещенных пероксидов ароилов, аддукты, отвечающие их присоединению к ФБН, характеризуются параметрами спектров ЭПР, близкими к таковым для аддуктов ФБН с бензоилоксильным радикалом [48,49,50] (αN =12,7 - 13,1, αo-H = 1,2 - 1,45).
Спиновые аддукты ФБН с фторсодержащими арильными радикалами, спектры ЭПР которых аналогичны аддуктам ФБН и замещенных фенильных радикалов [49,51] (αN=13,9-14,4, αo-H = 2,1-4.4), зафиксированы только для случаев разложения малофторированных пероксидов ароилов. Аналогичным образом при использовании в качестве спиновой ловушки нитрозодурола зафиксирован его аддукт только с п-фторфенильным радикалом, генерированным термолизом соответствующего пероксида п-фторбензоила [54]. В то же время есть данные, что при генерировании пентафторфенильного радикала из пентафторфенилазотрнфенилметана в ароматических субстратах в присутствии в качестве спиновых ловушек как ФБН [51], так и НД [52] удается зафиксировать соответствующие спиновые аддукты пентафторфенильного радикала.
Наиболее принципиальной и важной является фиксация аддуктов арилциклогекса-диенильных радикалов - интермедиатов радикальных реакций аренов. При использовании в качестве спиновой ловушки ФБН удается зафиксировать его аддукт с пентафторфенилциклогексадиенильным радикалом, образующимся при присоединении к бензолу .С6F5, генерированного как термолизом пероксида пентафторбензоила [54], так и из пентафторфенилазотрифенилметана [51]. При термолизе фторсодержащих пероксидов бензоила (кроме упомянутого выше пероксида п-фторбензоила) в бензоле в присутствии НД при 100-115°С образуются аддукты, в спектрах ЭПР которых отсутствует СТВ с ядром фтора, а константы СТВ αN, αo-H и αм-H близки к таковым для аддуктов п-замещенного фенильного радикала с НД. На этом основании было высказано предположение, что зафиксированы радикалы, формально являющиеся аддуктами п-арилзамещенных феиильных радикалов и НД, но получающиеся в результате окисления первоначально образующихся аддуктов арилциклогексадиенильных радикалов [54] (схема 21).
Аддукт (65a) (X = Н), наряду с его предшественником - аддуктом пентафторфенилциклогексадиенильного радикала (64a) (X=Н), (αN=13,2, αβ-H=9,3) - был зафиксирован при генерировании пентафторфенильного радикала из пентафторфенил-азотрифенилметана в бензоле в присутствии НД [52].
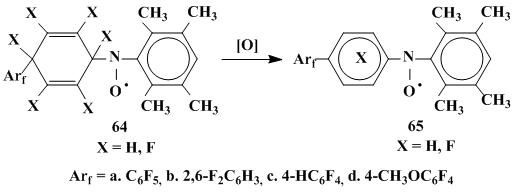
Схема 21
Для случаев атаки полифторированного ароматического субстрата арильными радикалами, генерированными из пероксидов ароилов, содержащих 4 и 5 атомов фтора, окисление образующихся первоначально арилгексафторциклогексадиенильных радикалов существенно затруднено. Это позволило зафиксировать спиновые аддукты соответствующих полифторарилгексафторциклогексадиенильных радикалов с НД 64a,c,d X=F [54].
Таким образом, использование метода спиновых ловушек для изучения реакций гексафторбензола с фторсодержащими арильными и ароилоксильными радикалами дает возможность фиксировать в виде соответствующих аддуктов не только атакующие ароматический субстрат радикалы, но и промежуточные частицы этих реакций - полифторированные циклогексадиенильные радикалы, что имеет большое значение для обоснования представлений об их участии в качестве интермедиатов радикальных реакций полифторароматических соединений.
Использование метода ЭПР для прямого наблюдения и идентификации циклогексадиенильных радикалов, образующихся в изученных нами реакциях полифторароматических соединений, рассмотрено при обсуждении обнаруженной нами перегруппировки полифторированных циклогексадиенильных радикалов с 1,2-миграцией атома фтора.
Наиболее характерные пути превращения полифторированных циклогексадиенильных радикалов, реализующиеся в ходе радикальных реакций полифтораренов, показаны на схеме 20. Только два из представленных на схеме путей аналогичны тому, что было показано для ипсо-реакций галоидароматических соединений: обратимое присоединение атакующего радикала и дегалоидирование с образованием ароматических соединений.
Обратимость присоединения радикала к субстрату при интерпретации ориентации в радикальных реакциях полифтораренов является весьма важным вопросом. Результаты изучения реакций полифторароматических соединений с алкильными радикалами дают основание считать, что присоединение таких радикалов, как метильный и циклогексильный, практически необратимо в широком интервале температур (25-140°С для .СН3 и 140-200°С для ц-.С6Н11) [13]. Присоединение трифторметильного радикала к гексафторбензолу становится обратимым только при температуре выше 180°С [55]. Сведений, касающихся перфторэтильного радикала, нет, однако, пользуясь сравнительными данными о температурах, при которых присоединение трифторметильного и перфторэтильного радикалов к бензолу становится обратимым (150°С для .СF3 [56, 57] и 110°С для .С2F5 [58]), можно предположить, что σ-комплекс перфторэтильного радикала с гексафторбензолом будет претерпевать возврат к исходным соединениям при более низкой температуре, чем соответствующий σ-комплекс трифторметильного радикала.
При изучении взаимодействия бензола и гексафторбензола с дифторхлорэтильным радикалом, генерированным фотолизом 1,3-дихлортетрафторацетона, обнаружены существенные различия в зависимости скорости присоединения .СF2С1 к этим субстратам от температуры, что связывается с различием температур, при которых становится существенной обратимость этого процесса [59]: для бензола - при 100°С, для гексафторбензола - при 250°С.
Несмотря на ограниченность информации, из сказанного выше можно заключить, что присоединение алкильных радикалов к гексафторбензолу становится обратимым при более высоких температурах, чем к бензолу.
Можно ожидать, что такая тенденция сохраняется и для присоединения к аренам арильных радикалов. Присоединение фенильного радикала, генерированного разными методами [30,60-66], к бензолу, несмотря на противоречивость данных о наличии в этих реакциях изотопного эффекта, считается обратимым [67] только при температуре порядка 200°С. В то же время присоединение пентафторфенильного радикала к гексафторбензолу в интервале 200-250°С необратимо [20,27,28]. Присоединение метильного и пентафторфенильного радикалов к октафторнафталину обратимо при более низкой температуре (140-160°С для первого и 200°С для второго случая [35]), чем для гексафторбензола, что в значительной степени определяет ориентацию в случае октафторнафталина.
В ряду гетероатомных радикалов, вовлекавшихся в реакции с полифтораренами, строгие доказательства обратимости присоединения имеются только для пентафторбензоилоксильного радикала [19,20,27,28,30,35,38,63-69].
На основании этих данных можно полагать, что основным фактором, влияющим на отрыв из геминального узла фрагмента, соответствующего присоединившемуся к субстрату радикалу, служит, по-видимому, температура, а не природа последнего.
Отрыв атома фтора из геминального узла полифторированного σ-комплекса является одним из путей образования конечного продукта. Роль дегалоидирующих агентов могут играть как сами ипсо-радикальные σ-комплексы, так и другие радикалы и компоненты реакционных смесей. Возможность ароматизации полифторированных циклогексадиенильных радикалов этим путем существенно зависит, таким образом, от наличия в реакционной среде реагентов, способных отрывать ипсо-атомы фтора (радикальные частицы, металлы и их галогениды, стенки реакционных сосудов, кислород воздуха и др.). В соответствии с этим в реакциях с алкильными радикалами, не содержащими атомов фтора, возможность ароматизации промежуточно образующихся радикальных σ-комплексов с превращением в продукт замещения атома фтора зависит от способа генерирования радикалов, определяющего в свою очередь присутствие различных компонент. Например, при генерировании метильного радикала из пероксида ацетила (80°С) выход продукта замещения фтора на метильную группу в гексафторбензоле не превышает 15-20% (основное направление реакции — рекомбинация σ-комплексов) [4]. В то же время дефторирование является практически единственным направлением превращения полифторированных циклогексадиенильных радикалов, образующихся в реакциях полифторированных соединений бензольного ряда и октафторнафталина с алкильными радикалами (.СН3 и .СН3СНОС2Н5), генерированными в системе СН3МgI - галогенид меди в диэтиловом эфире. В качестве эффективного дефторирующего агента в этих реакциях выступает, по-видимому, галогенид меди и восстановленный металл [12,10].
При фотолитическом методе генерирования алкильных радикалов наблюдается более эффективное, по сравнению с термическими реакциями, дефторирование радикальных σ-комплексов. Так, при термическом генерировании циклогексильного радикала даже при наличии в реакционном сосуде кислорода доля замещения атома фтора в гексафторбензоле [13] не превышает 20 % (в отсутствие кислорода воздуха основное направление реакции - рекомбинация σ-комплексов). При фотолизе же циклогексана в гексафторбензоле ароматизация σ-комплекса, отвечающего присоединению циклогексильного радикала к гексафторбензолу, является преимущественным направлением реакции (выход циклогексилпентафторбензола 61%) [70].
Вклад дефторирования циклогексадиенильных радикалов, образующихся в ходе взаимодействия полифторароматических субстратов с нефторированными арильными радикалами, генерированными из пероксидов ароилов, в суммарную картину взаимодействия колеблется в пределах от 30 до 80% для реакций фторированных соединений бензольного ряда [17,71] и от 15 до 45% - для октафторнафталина [36]. Было предположено, что эффективным дефторирующим агентом в этих реакциях является бензойная кислота. О том, что другим возможным участником процесса дефторирования радикальных σ-комллексов могут являться стенки реакционных сосудов, свидетельствует образование SiF4 в реакциях октафторнафталина с пероксидом бензоила как источником фенильного радикала [38,36].
В большинстве случаев взаимодействия полифторароматических соединений с полифторированными алкильными и арильными радикалами не реализуется гомолитическое замещение атома фтора, т. е. указанная выше ароматизация радикального σ-комплекса. Наличие в его геминальном узле атома фтора и сильно электроотрицательной полифторированной группы делает этот σ-комплекс стабильным по отношению к дефторированию. Известно возрастание прочности связи C-F с накоплением геминальных атомов фтора [72]. Есть также данные о более высокой стабильности полифторированных аренониевых ионов, имеющих два атома фтора в геминальном узле, по сравнению с изомерами, содержащими в геминальном узле отличные от атома фтора заместители [73]. Исключение составляет реакция октафторнафталина с таким источником пентафторфенильного радикала, как пентафторбензолсульфофторид (200°С), но выход перфторфенилнафталинов [35] и в этом случае не превышает 20% .
При взаимодействии полифтораренов с гетероатомными радикалами вклад дефторирования промежуточно образующихся радикальных σ-комплексов в общую совокупность их превращений также изменяется в широких пределах. Так, он составляет всего 2-10% для взаимодействия гексафторбензола и октафторнафталина с бензоилоксильным [17,36,71] и пентафторбензоилоксильным [18,20,38] радикалами, генерированными термически из соответствующих пероксидов ароилов, что, безусловно, связано с невысокой концентрацией этих радикалов, образование и превращения которых конкурируют с другими процессами, характерными для этих реакций (генерирование арильных радикалов и их реакции с полифтораренами). Преобладающим гомолитическое замещение (50-60%) становится для взаимодействия гексафторбензола с фотохимически генерируемыми кремнийсодержащими радикалами - триметил- и трихлорсилильным, которые сами выступают как дефторирующие агенты [74].
Фрагментация σ-комплекса с генерированием нового радикала является, по существу, ароматизацией с отрывом атома фтора, но в ситуации, когда его акцептором служит фрагмент геминального по отношению к нему заместителя (схема 22).
По-видимому, реализация такого процесса связана с определенными требованиями к легкости разрыва связи X-Y и, возможно, к повышенной прочности связи Y-F. Фрагментация такого типа имеет место, например, при взаимодействии октафторнафталина с кислородцентрированными радикалами [35,36,38,39].
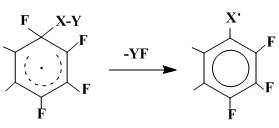
Схема 22
Для случая, когда заместитель в геминальном узле ХY = ArCOО, продукт фрагментации YF, представляющий собой фторангидрид соответствующей бензойной кислоты, был выделен. Другой продукт фрагментации радикального σ-комплекса - гептафторнафтоксильный радикал - далее превращается в оксопроизводные полифтордигидронафталинов и полифторнафтохиноны, являющиеся конечными продуктами этих реакций (схема 16).
Можно предположить, что фрагментация с генерированием нового радикала реализуется как основной путь стабилизации σ-комплексов, образующихся в результате присоединения к гексафторбензолу [6] .N02 или гидроксильного радикала [41,75]. Отщепление при этом NOF в первом случае и HF - во втором должно приводить к образованию пентафторфеноксильного радикала. В соответствии с этим из продуктов реакции в обоих случаях был выделен пентафторфенол и в случае с ОН с помощью УФ-спектроскопии установлено присутствие в продуктах реакции димеров гептафторфеноксильного радикала [75].
Рекомбинация, в том числе димеризация, является наиболее характерным путем превращения полифторированных циклогексадиенильных радикалов. Рекомбинация радикального σ-комплекса с исходным радикалом с образованием производного полифторированного циклогексадиена как основной путь стабилизации интермедиата наблюдается в реакции декафторбифенила с трифторметильным радикалом. Сопоставление этих данных с результатами взаимодействия гексафторбензола с трифторметильным радикалом, которое приводит главным образом к образованию продуктов димеризации σ-комплексов, отвечающих присоединению .СF3 к С6F6, дало основание связать возможность рекомбинации σ-комплексов присоединения .СF3 к С12F10 с относительно большим временем жизни циклогексадиенильного радикала в клетке обладающего высокой вязкостью декафторбифенила, в среде которого проводится реакция [16] (ср. [29]).
Рекомбинация радикальных σ-комплексов является основным направлением их стабилизации в тех случаях, когда ароматизация путем отщепления атома фтора затруднена из-за отсутствия эффективных дефторирующих агентов. Очевидно также, что из-за высокой прочности связи С-F рекомбинация оказывается более предпочтительной, чем диспропорционирование, которое, как и рекомбинация, требует участия двух σ-комнлексов. Продукты рекомбинации и димеризации радикальных полифторированных σ-комплексов образуются, как правило, с высокими выходами практически независимо от температуры и концентрации реагента, являющегося источником радикала. Например, в реакции С6F6 с (С6F5СОО)2 суммарные выходы этих продуктов достигают 90-98 % при изменении температуры проведения реакции от 80 до 200°С и концентрации пероксида [20] от 0,25 до 1 М. Исключение составляют те случаи, когда указанные факторы способствуют протеканию и конкурентных процессов. Так, при разложении пероксида перфторпропионила в гексафторбензоле [5] повышение температуры реакции от 50 до 80°С приводит к уменьшению выхода продуктов димеризации σ-комплексов, соответствующих присоединению .С2F5, почти в 2 раза, а увеличение концентрации пероксида в 25 раз (мольное отношение пероксид : гексафторбензол 1:40 и 1:1.5 соответственно) снижает выход димеров в 10 раз. В реакции гексафторбеизола с пероксидом ацетила [4] аналогичное увеличение концентрации пероксида приводит к практическому исчезновению из продуктов реакции димеров соответствующих σ-комплексов [3].
Имеются свидетельства того, что рекомбинация σ-комплексов в условиях этих реакций практически необратима. Например, длительное выдерживание продуктов взаимодействия полифторарена с источником радикала в условиях проведения соответствующей реакции не приводит ни к взаимным превращениям первично образующихся димеров σ-комплексов или продуктов рекомбинации последних с исходным радикалом или другими σ-комплексами, ни к появлению каких-либо других продуктов [3]. В известных к настоящему времени случаях гомолитический распад продуктов рекомбинации и димеризации на соответствующие σ-комплексы происходит, как правило, при температурах, намного превышающих те, при которых они образуются в результате радикальной атаки полифтораренов
Перегруппировка с 1 ,2-миграцией атома фтора от насыщенного атома углерода к соседнему радикальному центру является одним из наиболее важных путей стабилизации радикальных полифторированных σ-комплексов. Обнаруженная как первый пример изомеризации указанного типа в ряду свободных радикалов [19], она по своему значению выходит за рамки химии полифтораренов и имеет отношение к общей проблеме перегруппировок в результате миграции атома галогена, реализация которых еще в середине прошлого века считалась невозможной (см., например [76]). В 1962 г. появился первый обзор, в котором были обобщены данные об 1,2-миграциях атомов водорода и хлора, арильных и алкильных групп в свободных радикалах, протекающих в растворах [77]. В последующие годы экспериментальный материал, касающийся радикальных перегруппировок, постоянно наращивался и практически все обзоры, посвященные радикальным реакциям, включают рассмотрение перегруппировок свободных радикалов [78-81].
Ознакомление с этой литературой показывает, что перегруппировки радикалов, обусловленные миграцией атома галогена, до 1980 г. были известны главным образом для атома хлора [77,81,82] в полигалоидалкилах. Накопленный к настоящему времени экспериментальный материал свидетельствует о том, что 1,2-миграция атома хлора осуществляется внутримолекулярным путем [77], а результаты квантовых расчетов [83,84] показывают, что она идет через мостиковое переходное состояние с относительно невысоким энергетическим барьером (26,4 ккал/моль) [84]. Следует отметить, что во всех случаях, когда возможна конкуренция между 1,2-сдвигами атома хлора и фтора, наблюдается сдвиг атома хлора [85,86].
Миграция атома брома в ряду полигалоидалкильных радикалов протекает с большими скоростями, чем миграция хлора (1010 и 108 с-1 соответственно), поэтому она наблюдается реже [87]. Более предпочтительным в этом случае является отщепление брома, приводящее к образованию непредельных соединений.
Наиболее показательны примеры 1,2-миграции атомов брома в циклогексадиенильных радикалах. Так, анализ спектров ЭПР показывает, что возникающие при Х-облучении метанольных растворов бромбензола и п-бромфенола при 77 °К циклогексадиенильные радикалы с атомами брома в геминальном узле претерпевают быструю перегруппировку посредством 1,2-сдвига атома брома (время жизни зафиксированного состояния 10-7 с) [88].
Перегруппировка радикалов с 1,2-миграцией атома фтора, как уже отмечалось, впервые была обнаружена [19] и подробно исследованна [3] нами для полифторированных циклогексадиенильных радикалов. Однако, прежде чем обратиться к анализу закономерностей протекания этих превращений, имеет смысл коротко рассмотреть появившиеся позднее данные, касающиеся перегруппировок более простых фторсодержащих радикалов, протекающих с 1,2-миграцией атома фтора.
Так, была обнаружена миграция атома фтора в 1,2-дифторэтильном: радикале, образующемся в результате присоединения трития к транс- или цис-1,2-дифторэтилену [89].
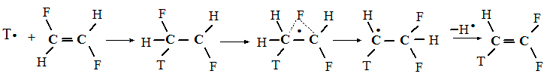
Японские химики [90] связали преимущественное образование СF2=СНТ при действии трития на 1,1,2-трифторэтилен с 1,2-миграцией атома фтора в первоначально образующемся радикале СF2СНТF, полагая, что разница в прочности связей С-F и С-Н не дает оснований считать, что возможен прямой межмолекулярный путь этого превращения, реализация которого требует предпочтительного отрыва атома фтора от указанного радикала. Анализируя с этих позиций неожиданный факт образования с высоким выходом СF3СН2СН2СF3 в результате присоединения атома водорода к трифторэтилену [91], японские исследователи [92] для объяснения этого результата также предложили 1,2-миграцию атома фтора в образующемся на первой стадии радикале СF2СН2F и последующую димеризацию продукта этой перегруппировки - радикала СF3СН2. Этими же авторами [92] определена константа скорости миграции атома фтора, равная 2. 1010 с-1.
Известно, что в этильном радикале 1,2-миграция атома водорода не имеет места [80], а энергия связи С-F во фторированных углеводородах ненамного больше, чем связи С-Н. Это явное несоответствие стимулировало поиск теоретических подходов к объяснению предпочтительности внутримолекулярной миграции атома фтора. Выполнены молекулярно-орбитальные расчеты энергетических барьеров 1,2-миграции атомов водорода, хлора [83,93,94] и фтора [83,95,96], которые свидетельствуют о высоком барьере такой миграции для водорода и фтора. В работе [97] с позиций теории возмущений была сделана оценка стабилизации β-галогенэтильных радикалов за счет мостикообразования:
-∆Е (ккал/моль): I (7,54), Вr (2,97), Сl (0,22), F (-1,30).
Эти данные говорят о невыгодности мостикообразования для атома фтора. Основной причиной этого, по мнению автора [83], является большая разность энергий взаимодействующих орбиталей, которая уменьшается при переходе от фтора к хлору, брому и йоду. В этом же направлении изменяется и легкость 1,2-миграции атома галогена.
С другой стороны, результаты расчетов методом INDO свидетельствуют о том, что высота барьера 1,2-миграции атома фтора в 1,2-дифтор- и 1,1,2-трифторэтильном радикалах [53], а также в 1,1,2,2-тетрафторэтильном радикале [98] через промежуточную мостиковую структуру более чем в 2 раза меньше, чем для аналогичной миграции атома водорода.
В зависимости от используемого метода расчета высота барьера 1,2-миграции атома фтора в 2-фторэтильном радикале изменяется от 28,6 до 107 ккал/моль [83,96], что показывает наличие определенных трудностей в теоретической интерпретации такой миграции. Более подробно перегруппировки с миграцией атома фтора и других галогенов рассмотрены в обзоре [99].
1,2-Миграция атома фтора в полифторированных циклогексадиенильных: радикалах, как показано нами [13] на основании анализа кинетической схемы реакции гексафторбензола с циклогексильным радикалом, является мономолекулярным процессом. Отношение выхода продуктов димеризации перегруппированных (П) и неперегруппированных (НП) σ-комплексов является линейной функцией концентрации радикала, зависящей от начальной концентрации его источника:
НП/П = К[R.] = f (Со источника радикала).
На примере взаимодействия гексафторбензола с фенильным, пентафторфенильным и циклогексильным радикалами было установлено, что повышение температуры и уменьшение концентрации источника радикала приводит к увеличению степени перегруппировки [3] (см., например, таблицу 2).
Оценка влияния на возможность протекания перегруппировки с 1,2-миграцией атома фтора стерических напряжений в геминальном узле полифторированных радикальных σ- комплексов, проведенная на примере реакций гексафторбензола с метильным и циклогексильным радикалами, близкими по полярности и заметно отличающимися по своим стерическим требованиям, показывает, что такое влияние незначительно [3].
Иллюстрируют этот вывод данные таблицы 3 по изменению степени перегруппировки (отношение выходов димеров перегруппированных и неперегруппированных σ-комплексов присоединения соответствующих радикалов к гексафторбензолу) в зависимости от концентрации источника радикала (.СН3 генерируется при 140°С из пероксида ди-трет-бутила, а .С6Н11 при 140°С из циклогексана в присутствии пероксида ди-трет-бутила) [3].
Таблица 3. Степень перегруппировки с 1,2-миграцией атома фтора в реакции гексафторбензола с алкнльными радикалами [3]
|
Концентрация пероксида |
Радикал |
|
|
СН3 |
ц-C6H11 |
|
|
0,77 0,42 0,38 0,33 0,19 |
0,29 0,50 0.58 0,85 1,12 |
0,27 0,32 0,47 0,58 0.68 |
Для оценки влияния природы заместителя в геминальном узле полифторированного радикального σ-комплекса на вероятность 1,2-миграции фтора из геминального узла был проведен расчет (расчет выполнен Л.Н.Щеголевой) неограниченным методом Хартри-Фока в приближении ЧПДП со стандартной параметризацией Попла и Сегала полных энергий и распределения спиновых плотностей в σ-комплексах, отвечающих присоединению к С6F6 метильного и трифторметильного радикалов, с одной стороны, и возникающих из них в результате 1,2-миграции атома фтора перегруппированных σ-комплексов - с другой [3].
Результаты расчета показывают, что для метильного радикала выгоднее на 11,6 ккал/моль σ-комплекс, отвечающий перегруппировке с миграцией атома фтора из геминального узла в соседнее положение, а для трифторметильного радикала картина обратная: на 3,8 ккал/моль выгоднее первоначально образующийся σ-комплекс с атомом фтора и трифторметильной группой в геминальном узле (схема 23).
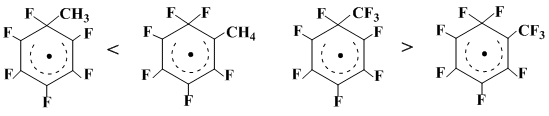
Схема 23
Недавно были опубликованы результаты теоретических исследований миграции атома фтора в свободных радикалах, в том числе полифторированных циклогексадиенильных радикалах. Результаты расчета показывают, что энергетический барьер перегруппировки (ΔE#)с 1,2-сдвигом атома фтора во фторированных циклогексадиенильных радикалах существенно ниже, чем в алкильных радикалах и осуществляется, вероятно, через мостиковое переходное состояние [100].
Таким образом, возможной движущей силой перегруппировки с 1,2-миграцией атома фтора может быть выигрыш в энергии при образовании геминального узла с двумя атомами фтора. Возрастание прочности связи С-F с накоплением геминальных атомов фтора известно [84]. Однако в тех случаях, когда заместителями в геминальном узле полифторированного радикального σ-комплекса являются атом фтора и группа с высокой электроотрицательностью (перфторалкильная или пентафторбензоилоксильная) и его электроноакцепторные свойства весьма сильны, миграции фтора к соседнему атому углерода не происходит.
Подтверждением этого служат полученные нами результаты прямого наблюдения методом ЭПР полифторированных циклогексадиенильных радикалов, образующихся в изученных радикальных реакциях полифтораренов [101].
Первые сведения о прямом наблюдении с помощью метода ЭПР циклогексадиенильных радикалов, образующихся в гомолитических реакциях полифторароматических соединений, были получены для радикалов, отвечающих присоединению атомов протия и дейтерия к гексафторбензолу и генерированных Х-облучеиием С6F6 соответственно в адамантановой и адамантан-d16-овой матрице [102].
Детектирование методом ЭПР гексафторциклогексадиенильных радикалов в растворе оказалось технически более простым, чем соответствующих водородсодержащих аналогов (интенсивность сигналов ЭПР фторсодержащих радикалов в сравнимых условиях намного больше, чем водородсодержащих), что позволило получить обширную информацию о структуре полифторированных циклогексадиенильных радикалов, константах скорости их образования в результате присоединения радикалов к гексафторбензолу [103]. Так, были зафиксированы полифторированные циклогексадиенильные радикалы (R.С6F6), генерированные фотохимическим разложением соответствующих источников атакующих радикалов R. в растворе С6F6 при 25 °С. Константы СТВ для зафиксированного этим способом радикала HС6F6., находятся в хорошем согласии с аналогичными характеристиками для того же радикала, генерированного в адамантановой матрице [102].
Природа радикала .R проявляется в спектрах ЭПР гексафторциклогексадиенильных радикалов, соответствующих его присоединению к С6F6, в величине константы СТВ для ипсо-атома фтора, которая изменяется от 65 Гс для .R = .ОС2Н5 до 152 Гс для .R = .Si(С2Н5)3 . Эти величины отражают стерические и электронные свойства радикала .R, а также дают информацию о структуре гексафторциклогексадиенильного фрагмента. Так, сравнивая закономерности изменения константы СТВ F1 в ряду гексафторциклогексадиенильных радикалов, отвечающих присоединению к С6F6 С-центрированных радикалов, Ингольд [103] предполагает, что имеет место внеплоскостное искажение циклогексадиенильного фрагмента, степень которого определяется, по-видимому, стерическими требованиями .R.
На примере значительного уменьшения величины константы СТВ с ядром 1Н при переходе от циклогексадиенильного или пентафторциклогексадиенильного радикала к гексафторциклогексадиенильному радикалу показано, что замена геминального атома водорода на атом фтора, которая вряд ли может вызвать существенные искажения плоской структуры циклогексадиенильного радикала, сопровождается эффектом, согласующимся с этим предположением [103]. Если приведенные соображения верны, то следует ожидать еще более низкого значения по сравнению с циклогексадиенильным радикалом для констант СТВ с ипсо-атомом фтора в гептафторциклогексадиенильном радикале. К сожалению, попытка осуществить фотохимическую реакцию присоединения атома фтора к гексафторбензолу оказалась безуспешной [103] и соответствующий гептафторциклогексадиенильный радикал с двумя геминальными атомами фтора не был зафиксирован.
Авторами работы [104] были получены спектры ЭПР циклогексадиенильных радикалов, образующихся в результате улавливания бензолом и гексафторбензолом ароилоксильных и арильных радикалов, генерированных прямым УФ-фотолизом (ртутная лампа высокого давления, 1000 W) диароилпероксидов в кювете ЭПР-спектрометра при 25 °С.
В случае гексафторбензола фиксируются спектры ЭПР циклогексадиенильных радикалов, отвечающих присоединению к субстрату как ароилоксильных, так и арильных радикалов.
В спектрах ЭПР циклогексадиенильных радикалов, отвечающих присоединению ароилоксильных радикалов к С6F6, константы СТВ с ипсо-атомом фтора имеют меньшие значения, чем в арилциклогексадиенильных радикалах. Причины этих различий, очевидно, те же, что и в случае рассмотренных выше гексафторциклогексадиенильных радикалов [103].
Нами [101] при нагревании (80оС) раствора пентафторбензоилпероксида в гексафторбензоле или при его фотохимическом разложении (20 оС) был зарегистрирован спектр ЭПР радикала, которому на основании сравнительного анализа констант СТВ (αF(1)=82.0; αF(2,6)=22,4; αF(3,5)=6,5; αF(4)=36,8), близких по значению известным константам для гексафторциклогексадиенильных радикалов с замещенными бензоилоксильными группами [103] была приписана структура перфтор-1-бензоилокси-циклогекса-диенильного радикала 18.
Поскольку при термическом разложении пентафторбензоилпероксида в гексафторбензоле перегруппировка с миграцией атома фтора наблюдается как вторичный процесс [19] при повышенной температуре, мы использовали в качестве субстрата перфтор-п-ксилол, опираясь на наши данные о том, что при разложении пентафторбензоилпероксида в октафтортолуоле [27] при 80-200оС в реакции участвует только пентафторфенильный радикал.
При нагревании (90-100оС) раствора пентафторбензоилпероксида в перфтор-п-ксилоле фиксируется спектр ЭПР 1-пентафторфенил-3,6-бистрифторметил-2,4,5-трифторцикло-гексадиенильного радикала (66). Величина αF(1)=69.2 для радикала 66 меньше соответствующей константы как для радикала 18 (αF(1)=82.0), так и фенилгексафторциклогексадиенильного радикала [103] (αF(1)=107.3). Это может быть результатом влияния не только высокой электроноакцепторности пентафторфенильной группы в геминальном узле радикала 66, но и стерического взаимодействия с CF3 группой во 2 положении циклогексадиенильного кольца. При этом возможный вывод геминального узла из плоскости и искажение плоскости π-системы циклогексадиенильного фрагмента приводят к отклонению величин αF(3), αF(4), и αF(6) радикала 66 от типичных для незамещенных полифторированных циклогексадиенильных радикалов констант СТВ [103,104].
При повышении температуры реакции до 120-138 оС в спектре ЭПР радикала 66 наблюдаются заметные изменения, свидетельствующие о генерировании нового радикала (67). В спектре появляются две константы СТВ αF(1)(50.4 и 50.0) с близкими значениями, что указывает на изомеризацию радикала 66 с миграцией атома фтора из геминального узла к соседнему атому углерода положение, занятое атомом фтора, то есть радикал 67 имеет структуру перфтор-2,5-диметил-6-фенилциклогексадиенильного радикала. Меньшие значения констант СТВ αF(1) радикала 67 по сравнению с радикалами с геминальным узлом CFR согласуются с известной зависимостью величин αF(1) от стерических взаимодействий заместителя в геминальном узле и его электроотрицательности [103]. Наличие двух констант СТВ αF(1) свидетельсьвует о неэквивалентности атомов фтора геминального узла, что может быть связано с его выводом из плоскости молекулы. Однако близкие значения этих констант указывают на то, что искажение это незначительно и не влияет на плоское строение циклогексадиенильного фрагмента радикала 67. Величина g=2.0037±0.0003 этого радикала типична для радикалов такого строения [103], а константы СТВ αF(3)=6.5 и αF(4)= 38.2, в отличие от предшественника этого радикала 66, близки к значениям для гексафторциклогексадиенильных радикалов, замещенных только в геминальном узле [103].
Дополнительным подтверждением структуры радикала 67 является его генерирование при нагревании (100-130 оС) димера (68), выделенного препаративно [101] из продуктов разложения пентафторбензоилпероксида в перфтор-п-ксилоле при 140 оС. (схема 24).
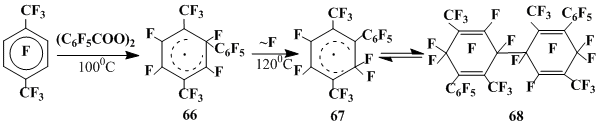
Схема 24
Заключение
В заключение следует назвать наиболее важные особенности поведения полифторароматических соединений в радикальных реакциях:
1.Предпочтительная димеризация радикальных σ-комплексов с образованием фторированных тетрагидробиарилов в отсутствие дефторирующих агентов, роль которых могут играть как специальные реагенты, так и продукты реакций.
2. Пониженная, по сравнению с не фторированными аналогами, реакционность полифторароматических соединений по отношению к электрофильным радикалам и повышенная - по отношению к нуклеофильным радикалам.
3. Предпочтительная мета-ориентация в реакциях соединений C6F5X (X=CF3, C6F5) с электрофильными радикалами (.CF3, .C6F5), и пара-ориентация в случае нуклеофильных радикалов (.CH3, .CH(CH3)OC2H5)
4. Для октафторнафталина - предпочтительная атака в 1-положение в реакциях с кислородцентрированными и алкильными радикалами при низкой температуре, и предпочтительное образование продуктов атаки в положение 2 алкильных и арильных радикалов при высокой температуре.
5. Неизвестная ранее перегруппировка с 1,2-миграцией атома фтора для полифторированных циклогексадиенильных радикалов - короткоживущих интермедиатов радикальных реакций (σ-комплексов)
6. Новый тип стабилизации радикальных σ-комплексов в результате их фрагментации до соответствующих ароксильных радикалов, как специфическая особенность реакций полифторароматических соединений с кислородцентрированными радикалами.
Список литературы
Материал рекомендован к публикации членом редколлегии В.Е. Платоновым
Fluorine Notes, 2012, 81, 1-2


