Fluorine Notes, 2000, 8, 5-6
|
 -Difluoroalkylamines:
2-chloro-1,1,2-trifluoro-N,N-diethyl-ethanamine and 1,1,2,3,3,3-hexafluoro-N,N-diethyl-propanamine
as fluorinating agents
-Difluoroalkylamines:
2-chloro-1,1,2-trifluoro-N,N-diethyl-ethanamine and 1,1,2,3,3,3-hexafluoro-N,N-diethyl-propanamine
as fluorinating agents |
S. Igoumnov and V. Kornilov |
|

1. 2-chloro-1,1,2-trifluoro-N,N-diethylethanamine (FAR)
Its other names:
Diethyl(1,1,2-trifluoro-2-chloroethyl)amine, N(2-chloro-2-fluoro-1,1-difluoroethyl)diethylamine, 2-chloro-1,1,2-trifluorotriethylamine, fluoroalkylamine reagent abbreviated as FAR, Yarovenko Reagent.
The main constants of FAR:
Boiling point: bp5,5-6 32-33oC[1,2], bp8 28-30oC [3],bp10 37o[4]
d4 25 1.19 [1].
FAR is extremely easily oxidized even with air moisture, it is kept in a close vessel in a desiccator without light access [5].
Due to high reactivity of fluorine atoms caused by influence of the electron-donating diethylamine group, FAR has been widely applied in preparative organic chemistry [6]. FAR instability during storage puts obstacles in the way of its industrial use [5].
1.1. Methods to produce FAR
In the late 40-s a scope of authors [1] studied reactions of polyfluoroolefins with
primary and secondary amines including reactions of chlorotrifluoroethylene with
diethylamine to form tertiary amine with 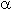 -difluoro structure. The reaction runs rapidly
with heat evolution:
-difluoro structure. The reaction runs rapidly
with heat evolution:
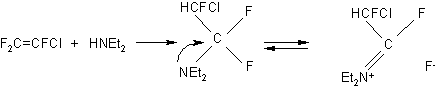
Substitution of the vinyl fluorine atom right up to the formation of tetrakis(dialkylamino)ethylene occurs in the amine excess under certain conditions. The synthesis is carried out in two ways:
- the starting compounds are charged into a pressure vessel( preliminarily cooled [7-10] or placed in a water bath for heat elimination [1]), kept for heating up to room temperature at which the reaction takes place. The product produced is subjected to vacuum distillation. The FAR yield is 80-90%.
- Chlorotrifluoroethylene is bubbled through a layer of diethylamine at cooling from –20 oC to +5oC[3]; from –10oC /–5oC [2,11,13] to –76oC [12], and in[14] it was mentioned that the reaction passes especially smoothly if a small amount of FAR is added into the starting diethylamine.
This route of the process leads to a yield of FAR of 61% [2], 44% [3], 79% [12], 84% [13] after vacuum distillation. Though the bubbling results in a lower yield of FAR, this process is more simple in technology because does not need pressurization [5].
1.2. Reactions using FAR
Though FAR and its reactions with alcohols, water and amines have been known [1],only in the late 50-s N.Yarovenko and M.Rakscha proposed to use FAR in reactions of substitution of hydroxyl and sulfohydryl groups with fluorine.
The reaction takes place according to the following scheme:
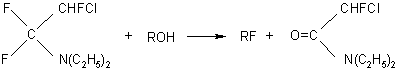
to form fluoride and side NN-diethyl-2-fluoroacetamide which is always formed in all reactions with use of FAR [15].The acetamide produced as a result of hydrolysis or alcoholysis of FAR is the starting compound for producing NN-diethyldifluoroacetamide {15,16,17].
1.2.1. Fluorination of steroid alcohols.
In the 60-70s FAR was used especially wide in producing novel steroids. It was suggested
that the fluorine atom in a specific site in the steroid molecule could intensify
pharmacological effects. A detailed review devoted to the production of steroids
with the use of FAR including publications from 1962 till 1971 was presented
in [5,pp 260-262,294-323]. After 1972 a number of methods to produce 11
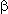 - Halosteroids, belonging to the estrane group, from their hydroxyl-containing
precursors was patented.
- Halosteroids, belonging to the estrane group, from their hydroxyl-containing
precursors was patented.
The reaction was carried out under different conditions:
- in CH 2Cl 2 solvent at a temperature of –15/-65oC [18]
- in THF at 0oC [19]
- in the presence of LiCl or LiF, here chlorosteroids are formed in the presence of LiCl and the presence of LiF results in formation of fluorosteroids [21].Allenyl steroids were produced also. In an intermediate stage, allenic acid fluorides [23] are formed which were then converted to b-oxo-esters;
- fluoro unsaturated steroids [24] were produced in a medium of different solvents (THF,CH 2CL 2,MeCN,CHCl 3)
1.2.2. Interaction of FAR with aliphatic compounds
To study reactions with the use of FAR, a wide scope of hydroxyl-containing compounds containing different functional groups (besides –OH) and different substituents was used including the following as the starting compounds:
a) halogenalkanols
- bromobutanols
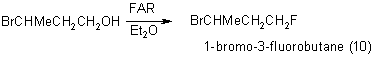
- 4-chloro-3,3,4-trifluoro-2-methyl-1-butanol forms with FAR products of –OH group substitution with fluorine or chlorine to get 1,2,2,4-tetrafluoro-3-methylbutane and 1,4-dichloro-1,2,2-trifluoro-3-methylbutane in a ratio of 7:3 [25]
- 2 Br(or I)-propan-1-ol reacts with FAR to form rearranged 1-bromo-2-fluoro derivatives, but 2-chloropropan-1-ol gives the product of direct substitution [26]
- paper [27] presents the results of investigation on effect of the reactor material on the yield of fluorides at interaction of FAR with fluoroalcanols. In a reactor of platinum the yield is higher than that in a glass reactor.
- The study of interaction of halogenalkylalcanols with FAR has shown that primary, secondary and tertiary alcohols containing fluoro(fluorochloro)alkyl groups in the a-position relative to hydroxyl do not react with FAR at room temperature with substitution of –OH with fluorine. At a temperature above 150oC a product of substitution by fluorine and chlorine is formed in a ratio of 25:75, for example, 2,2,3,3-tetrafluoropropanol with FAR gives 2 products, pentafluoropropane and chlorofluoropropane (20:80) [28]
b) Diols interact with FAR with the formation of cyclic compounds.
Ethyleneglycol forms chlorofluoroacetic acid orthoesters
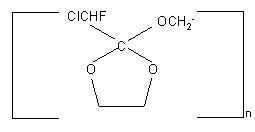 where n=2
where n=2
And 1,4- butandiol forms tetrahydrofurane [25] .
c) Substitution of –OH with F in optical active compounds is followed by inversion of configuration. Thus, (+)-s-2octanol forms (-)-R-2-fluorooctanol with 88% optical purity [29], and Me-1-acetyl-trans-4-hydroxy-L-prolinate was converted to Me-1-acetyl-cis-4-fluoro-L-proline with the use of FAR [30]
d)Carbohydrates of sugars type react with FAR to form chlorofluoroacetates in high yield. At more rough conditions, fluorination to a very small degree is observed [31,32,70]. Replacement of –OH group by fluorine occurs only at interaction of FAR with a-D-glucofuranose [33,34]
e) Replacement of hydroxyl with fluorine by means of FAR was used in an intermediate stage of producing fluoroglutamic acid and fluorofolic acid [35]
f) Carboxylic acids readily exchange hydroxyl with fluorine with the formation of acylfluorides in a yield over 75% [36,37]. But salts of carboxylic acids react with FAR only at heating.
g) Fluorodimethyl ether and fluoromethyl ether were produced by substitution of hydroxyl with fluorine in CF3CH(OH)OC2H5 and CF3CH(OH)OCH3 compounds [38,39,40]
1.2.3. Interaction of FAR with aromatic compounds.
Interaction of FAR with aromatic compounds containing carbonyl group has been described in [14]. Aldehydes of the following general formula
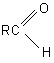
form RCF2H (R=C6H5 meta- and para-O2NC6H4) compound and diethylamide of fluorochloroacetic acid with FAR.
In 4-O2NC6H4O(CH2)3OH the hydroxyl is replaced by fluorine at FAR treatment [41].
A possibility to modify properties of substituted phenylethylamines, which are anorexigues and stimulators of central nervous system, by means of hydroxyl replacement by fluorine is of scientific and practical interest:
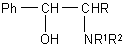
(R=H,Me,Ph
NR1R2 = -piperidino, -morpholino; -NMe2)
The reaction of the mentioned compound with FAR is stereospecific, it proceeds with configuration inversion and allows to get optically pure substance [42].
FAR is used to produce -fluorophenylaceto- and - fluorophenylpropionitriles from starting hydroxynitriles [43].
Substitution of –OH group in haloidhydrines or aminoalcohols leads to the formation of the appropriate fluorine-containing compounds [44].
At the same time FAR is an effective ring-closing reagent for ortho-bifunctional benzenes such as, for example, 2-aminophenol, ortho-phenylenediamine, 2-aminothiophenol, 2-aminobenzamide and catechol. As a result of this reaction, different benzoheterocyclic compound containing chlorofluoromethyl group are formed:
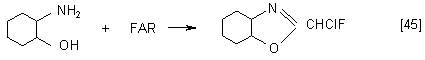
but in the presence of KF in a DMF medium, benzoxazole with difluoromethyl group is formed:
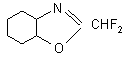
Paper [47] describes a simple route to introduce a-fluorinated carbonyl group into a molecule of an aromatic compound. FAR with BF 3 gives salts similar to ammonium salts which are able to acidify aromatic and heterocyclic compounds with elevated ring electron density (electron-rich aromatic compounds).
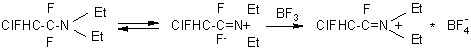

FAR enters the reaction with amides of arensulfo acids in the presence of KF to give enamines:
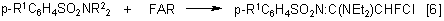
where R1=Me,Cl; R2 =H, Cl.
1.2.4. Interaction of FAR with cyclic compounds of different structure.
In compounds containing –OH group, replacement of this group by fluorine also takes place, for example, 4-t-butylcyclohexanol with FAR gives cis and trans 4-t-butylcyclohexyl fluorides [11].
Fluorobicyclo[2,2,2]octanes are formed from their hydroxyl-containing precursors in a 50-60% yield [48,49].
Interaction of FAR with adamantane carboxylic acids containing –OH and –CH2OH groups [50] and with hydroximethyloxazolidines [51] proceeds similarly.
A number of investigations carried out at the University of Leeds [52-56] has been devoted to synthesis of novel biologically active compounds, Halogibberellins. The conditions to produce halogenated gibberellins have been found.
Interaction of tetrahydrogibberellinic acid with FAR and LiX (where X=F,Cl,Br) in dimethoxyethane or THF results in formation of F-, Cl- or Br-substituted gibberellin[55,56].
Substitution of –OH with fluorine by means of FAR was also used:
- at a plant producing antibiotics of Halocephalosporins class [57,58]
- in production of fluorine-substituted morphine and codeine [59]
- in production of 7-fluoronorbornadiene [71]
1.2.5.Interaction of FAR with silanes
FAR gives a possibility to produce fluoropropylsilanes from hydroxypropylsilanes. Exchange of –OH for fluorine is accompanied by formation of cyclopropane and trimethylfluorosilane. The choice of reaction conditions, use of a low-boiling solvent allow to control partially this side reaction [12].
Production of trimethylfluorosilane has been described in [60]. The starting material is trimethylsilane Me 3SiX ( where X =Cl, OEt, OAc, ONa, O(CH2)6Me, OSiMe3) which with FAR gives trimethylfluorosilane in a 69-99% yield.
Reactions of FAR with other organic silicones were studied also.
Trimethylfluorosilane [61] is formed at interaction of trimethylsiloxypentachloro - 2-propene with FAR.
Trimethylsylilisocyanate with FAR in a ratio of 2:1 forms adduct which is easily decomposed to the starting materials and at a temperature from 80 to 100oC to trimethylfluorosilane and gumlike product [62].
N-trimethylsylilethanimine readily reacts with FAR at 30-40oC with formation of trimethylfluorosilane and unstable amine decomposing with formation of azopentadiene [63].
1.2.6. Miscellaneous
FAR enters a reaction with HCN to form HCFClC(CN) 2NEt 2 [64]. A mixture of methyliodide and silver borofluoride easily alkylate FAR to form quarternary ammonium compounds: [65].
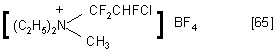
N-alkylidene-carbomoylisocyanates are fluorinated by FAR very easily according to nucleophilic mechanism with formation of a-fluoroalkylisocyanates which are readily hydrolysable [66].
In a similar way FAR reacts with compounds containing active methylene groups, malononitrile and cyanoacetic ether to form enamines. The reaction takes place in the presence of KF [6].
FBr used in situ in production of steroids is produced by BrNAc treatment with FAR [67].
Spiro[androstenedioxanes] with different substituents were produced by cyclization of hydroxy preynadienediones at their interaction with FAR [68].
Methioxy oxime was taken as example to show possibility of using FAR for Beckmann rearrangement
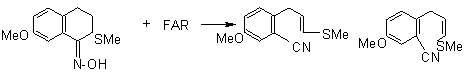
2. 1,1,2,3,3,3,-hexafluoro-N,N-diethyl-propanamine (PPDA)
Other names:
1) N(1,1,2,3,3,3,-hexafluoropropyl)diethylamine
2) Mixture of b-hydroperfluoropropyldiethylamine and perfluoropropenyldiethylamine
3) Adduct of perfluoropropene and diethylamine
4) Abbreviated as PPDA
5) Ishikawa Reagent
Main constants:
Bp52-52.5oC (55 mm)
nd20 1.3550; d420 1.2279 [72]
The results of investigation of interaction of perfluoroisobutene and perfluoropropene with nucleophilic reagents including interaction of perfluoropropene with diethylamine were reported in 1956 in paper [72]. The reaction was carried out in a medium of absolute ether at vigorous stirring. After completion of the reaction, the mixture was filtered from residue, ether was evaporated and the residue was distilled in vacuum. The analysis of the substance produced has shown that substitution of vinyl fluorine atom takes place together with amine addition to the double bond and the product produced is a mixture of the following amines: CF3CF=CF-N(C2H5)2 and CF3CFHCF2-N(C2H5)2.
Amines and olefins which one can use to produce compounds similar to FAR have been mentioned in [9], including PPDA.
In 1979 N.Ishkawa et al. suggested to use PPDA instead of FAR for fluorination of alcohols and carboxylic acids. The authors believe that it is more easy to produce PPDA and it is more stable than FAR [73].
The following different alcohols have been fluorinated with the use of PPDA:
a) lower primary alcohols with production of fluorides;
b) lower secondary and tertiary alcohols to form fluorides and olefins [73], in which connection reactions of dehydration, isomerization and dimerization of alcohols take place at the interaction of PPDA with most secondary alcohols, that leads to a low yield of fluorides [74];
c) higher primary fatty alcohols with PPDA form a mixture of fluorides and esters used as insecticides and surfactants. For example, lauryl alcohols with PPDA form lauryl-2,3,3,3-tetrafluoropropionate and lauryl fluoride.
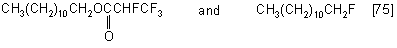
d) diols react with PPDA with the formation of cyclic adducts being 1,3-dioxane derivatives, a high yield is attained only when the reaction is carried out in a THF medium [76].
e) novel cyclic compounds, for example 3,9 bis(diethylamino)-3,9 bis(1,2,2,2-tetrafluoroethyl)-2,4,8,10-tetraoxaspiro[5,5]-undecane [76], were produced at interaction of glycerol or pentaeritritol with PPDA.
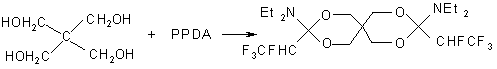
f) di-substituted glycerols, for example 2,3-dibromo-1-propanol with PPDA form ester-2,3-dibromo-propyl-2,3,3,3-tetrafluoropropionate, while mono-substituted glycerols and PPDA form cyclic adducts, 1,3-dioxalan derivatives [77].
g) hydroxyl in aliphatic and aromatic halogeno alcohols is replaced at interaction of PPDA with fluorine, but with cycloalcohols there are formed esters-tetrafluoropropionates. Thus, 3-bromoborneol and PPDA form 3-bromobornyl-2,3,3,3-tetrafluoropropionate [78];
h) Nitro alcohol, aromatic and aliphatic ones react with PPDA in different ways, particularly nitrobenzyl alcohols form nitrobenzyl fluorides, while aliphatic alcohols, for example 2-methyl-2-nitro-1-propanol, form 2,3,3,3-tetrafluoropropionates [79].
i) , - unsaturated alcohols at direct interaction with PPDA undergo a series of side
transformations such as isomerization, cyclization and dehydration. It has been
found that the cause of isomerization is HF forming during the process. For its
removal, the process is carried out in the presence of tertiary amine, diisopropylethylamine
for example. In this case esters-2,3,3,3-tetrafluoropropionate are formed [80].
Thus, interaction of geranoil with PPDA in the presence of the mentioned amine
results in the formation of geranyl-2,3,3,3-tetrafluoropropionate in 41% yield
[80].
k) PPDA is used to fluorinate steroid alcohols also [81];
j) aromatic hydroxyesters at interaction with PPDA form appropriate secondary fluorides in a 60-75% yield [74].
Reactions of PPDA with various fatty acids monoesters of ethylene glycol have been also investigated. Here a mixture of appropriate 2,3,3,3-tetrafluoropropionates as main products and monofluorides is formed. For example ethylene glucol monolaurate with PPDA forms fluoride in 3% yield and diester in 43.8% yield.
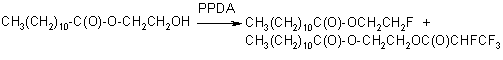
Diesters of ethylene glucol and ethylene glucol are used as antimicrobial reagents or plasticizers.
Interaction of PPDA with monoesters of aromatic carboxylic acids also leads to the formation of a mixture of fluorides and tetrafluoropropionates. Here in all cases N,N-diethyl-2,3,3,3-tetrafluoropropioamide, a side substance, is formed [82]. The use of PPDA in organic synthesis is not limited only to substitution of –OH group.
PPDA leads to heterocyclization of bifunctional benzene and haphtalene to produce tetrafluoroethyle benzo- and haphto-heterocycles.
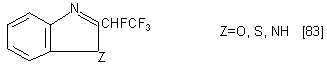
PPDA with PhNH2 forms the following unsaturated compound
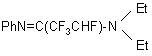
in 76% yield. In the starting compound, RNH 2, R may be alkyl, aryl, heterocycle.
Aetylenic ketones are formed when - diketones react with PPDA and freeze-dried KF. So, ketones of the formula

(R=Ph, R’=MeCO,F 3CCO,F 3CCF 2CO; R=4-ClC 6H 4, R’=F 3CCO;
R=Me, R’= MeCO, EtO 2C; R=Ph,R’= CO 2Et)
are formed in one stage in a 48-76% yield by treatment of RCOCH 2R’ with PPDA or FAR in the presence of KF [85].
PPDA is used in synthesis of fluorofenicol and its analogues from oxazolines [86]
and also in synthesis of pyretroids 10,10-difluorobiophenothrin and 7'-fluorobioallethrin
from oxidized derivatives of the parent insecticides [87]. Paper [88] describes
interaction of PPDA with  - hydroxy-, - unsaturated sulfones. In this reaction, -OH group
is not substituted , -lactone
is formed and only S-diastereomer with enantioselletivity of 99%.
- hydroxy-, - unsaturated sulfones. In this reaction, -OH group
is not substituted , -lactone
is formed and only S-diastereomer with enantioselletivity of 99%.
Conclusions
The use of FAR (Yarovenko agent) in developments began from the early 60-s and PPDA (Ishikawa reagent) from the early 80-s. The interest to these reagents was caused by a possibility to produce biologically active substances in a simple way. But due to instability of FAR and PPDA during storage, their use is limited to preparative methods.
Depending on the nature of a substance entering a reaction with FAR or PPDA, replacement of hydroxyle by fluorine takes place or formation of ether-fluoroacetates (FAR) or fluropropionates (PPDA) occurs. Bifunctional compounds give heterocyclic compounds with them. But cases of formation of oleophilic compounds, lactones etc. take place also.
References
- R.L. Pruett, I.T. Barr, K.E. Rapp and others., J. Amer. Chem. Soc., 1950, v.72, p.3646
- N.N. Yarovenko, M.A. Raksha., Zh. Obshch. Khim., 1959, v.29, p.2159
- L.H. Knox, E. Levarde, S. Beryer and others., J. Org. Chem., 1964, v.29, n.8, p.2187
- D.C. England, L.R. Melby, M.A. Dietrich., J.Org.Chem.Soc.,1960, v.82, p.5116
- C.M. Sharts, W.A. Sheppard., Org. Reactions,1974, v.21, p.158
- V.I. Pasternak, M.I. Dronkina, V.P. Kukhar, L.M. Yagopol'skii., Zh. Org. Khim.,1978, v.14, n.12, p.2493
- US Pat. 3118894, 1962-1964, CA,1964, 60, 12072
- US Pat. 3153644, 1962-1964, CA,1965, 62, 619
- US Pat. 3105078, 1962-1963, CA,1964, 60, 427
- M. Hudlicky, I. Lejhacova., Collect. Czech. Chem. Commun., 1966, v.31, n.3, p.1416, CA,1966, 64, 17404
- E.L. Elill, R.I. Martin., J.Amer.Chem.Soc.,1968, v.90, p.682
- V.B. Pukhnarevich, I. Vcelak, M.G. Voronkov.,Collect. Czech. Chem. Commun., 1974, v.39, n.9, p.2616
- F. Liska., Chem. Listy, 1972, v.66, n.2, p.189
- A.V. Fokin, V.I. Zimin and others., Zh. Obshch. Khim.,1968, v.38, n.7, p.1510
- V. Tolman., Collect. Czech. Chem. Commun.,1977, v.42, n.8, p.2537
- F. Liska, F.Hampl,V.Dedek., Collect. Czech. Chem. Commun., 1980, v.45, n.3
- F. Liska, F. Hampl, V. Dedek., CS Pat. 187198, 1977-81, CA,1981, 96, 34610
- NL Appl. 71 02079, 1971-1972, N.V. Organon, CA,1972, 77, 164977
- NL Appl. 72 09837, 1972-1974, AKZO N.V.,CA,1974, 81, 13707
- SA Pat. 7200618, 1972, N.V. Organon, CA,1972, 78, 136532
- US Pat. 3767685, 1972-1973, Akzona Inc.
- NL Appl. 72 09299 1972-1974, AKZO N.V.
- P. Crabbe, H.Caprio, E. Velarde., J.Org.Chem., 1973, v.38, n.8, p.1478
- A. Neder, A. Uskert, E. Nagy., Acta. Chim. Acad. Sc. Hung., 1980, v.103, n.3
- V. Dedek, F. Liska, L. Cvak., Collect. Czech. Chem. Commun., 1978, 43(10), 2649
- B.W. Bycroft, A. Chowdhury, M.A. Dove., J. Fluor. Chem., 1978, 12, n.2, p.45
- V. Dedek, F. Liska, L. Cvak., Collect. Czech. Chem. Commun., 1978, v.43, n.4, 985
- F. Liska, V. Dedek, Z. Chvatal , L.Cvak., Collect. Czech. Chem. Commun., 1975, v.40, p.1441
- J. Leroy, E. Hebert, C. Wakselman., J. Org. Chem. ,1979, v.44, n.19, 3406
- M. Hudlicky., J. Fluor. Chem., 1993, 60, p.193
- P.W. Kent .,Chem. and Ind., 1969, p.1128
- K.R. Wood, D. Fisher, P.W. Kent ., J. C. S. "С" Org., 1966, 21, p.1994, CA, 1967, 66, 11134
- L. Evelyn, L.D. Hall., Carbonyd. r. Res.., 1976 ,47, n.2, p.285, CA, 1976, 84,180492
- L. Evelyn, L.D. Hall., Chem. Ind. (London) ,1968, 6, p.183
- E.D. Berymann, Chun-Hsu Lin., Synthesis, 1973, 1, p.44, CA, 1973, 78, 111726
- K. Schaumbery., J. Magn. Resonance, 1972, 7, n.2, p.177, CA 1972, 72, 74359
- A.V. Fokin, Yu.N. Studnev and others., Izv. Akad. Nauk SSSR, Ser. Khim., 1984, 2, p.411
- R.D. Bagnell, W. Bell, K. Pearson ., J. Fluor. Chem., 1973, 13, n.2, p.123
- G. Siegemund .,Chem. Ber., 1973, 106, n.9, 2960, CA, 1973, 80, 3031
- G. Siegemund, R. Muschaweck., DE Appl. 2340560, 1973-1975, Farbwerke Hoechst AG, CA, 1975, 82, 155327
- JP Appl., 81 20549, 1979-1981, CA, 1981, 95, 42639
- S. Hamman, C.G. Beguin, C. Charlon., J. Fluor. Chem., 1987, 37, n.3, p.343
- C. Florin, J. Chantegler, C. Charlon and others., Ann. Pharm. Fr., 1985, 43, n.6, p.595, CA, 1987, 106, 138037
- C. Charlon, C.Lun-Duc., Ann. Pharm. Fr., 1986, 44, n.2, p.123, CA, 1987, 106, 66822
- A. Takaoka, K. Iwamoto, N. Ishikawa and others., J. Fluor. Chem., 1979, 14, n.5, p.421
- F. Liska, J. Vayner, V. Dedek ., Sb. Vys. Sk. Chem-Technol. Prazl. Org. Chem. Technol., 1986, 29, 37, CA,1989, 110, 57553
- C. Wakselman, M. Tordeux., J. Chem. Soc. ,Chem. Commun.,1975, n.23, p.956
- J. Kopecky, J. Smejkal, M. Hudlicky., Chem. and Ind., 1969, p. 271
- J. Kopecky, J. Smejkal., Collect. Czech. Chem. Commun., 1980, 45, n.11, p.2971
- A.M. Aleksandrov, C.J. Danilenko, L.M. Yagupolsky., Zh. Org. Khim., 1973, 9, n.5, p.951
- D.P. Schumacher, US Pat. 4876352, 1988-1989, Schering Corp., CA, 1989, 112, 198364
- J.H. Bateson, B.E. Cross., Tetr. Letters, 1973, n.2, p.1783
- J.H. Bateson, B.E. Cross., J.Chem. Soc. Perkin Trans. I, 1974, n.20, p.2409
- B.E. Cross, J.C. Simpson., Tetrahedron Lett., 1980, 21, n.2, p.215
- B.E. Cross, R.E. Banks., J.Chem. Soc. Perkin Trans.I, 1977, 5, p.512
- B.E. Cross J.C. Simpson., J.Chem. Soc. Perkin Trans.I, 1982, 11, p.2571
- R.R. Chavvette., DE Appl 2408686, 1973-1974. Lilly Eli and Co., CA, 1974, 82, 4277
- P. Schneider, H. Bickel., Helv. Chim. Acta., 1975, 58, n.8, p.2669, CA, 1976,84,121746
- D.E. Ayer, US Pat. 3137701, 1962-1964, CA, 1964, 61, 4409
- J. Vcelak, V. Chvalovsky., Synth. React. Inorg. Met-Org Chem., 1977, 7, n.2, p.123, CA, 1977 87, 135531
- V.P. Kuhar, I.V. Migaychuk, L.A. Lazutkina and others., Zh. Obshch. Khim., 1983, 53, n.7, p.1578
- M.N. Chertsyuk, M.I. Dronkina and others., Zh. Org. Khim., 1980, 16, n.7, p.1429
- V.I. Pasternak, M.L. Dronkina and others., Zh.Obshch. Khim., 1984, 54, n.5, p.1110
- L.R. Melby, D.C. Enyland., J.Amer.Chem.Soc.,1960, p.5116
- L.M. Yagupolsky, N.V. Kondratenko, M.I. Dronkina., Zh. Org. Khim.,1980, 16, n.12, p.2508
- M.N. Gertsyuk, V.L. Gorbatenko, M.I. Dronkina and others., Zh. Org. Khim., 1979, 15, n.7, p.1556
- R. Mickova, J. Moural, V. Schwarz., Tetrahedron Lett., 1978(15), 1315
- G. Anner, H. Kaufmann., DE Appl, 2451777, CA, 1975, 83, 97727
- R.L. Autrey, P.W. Scullard, J. Amer. Chem. Soc., 1968, 90, p.4924
- R.Zeitig, D.Cech, F. Liska., Z. Chem., 1983, 23, n.10, p.375, CA, 1984, 100, 139492
- M. Frank-Neumann, M. Sedrati., Bull. Soc. Chim. Fr., 1976 (9-10 Pt2), 1476, CA, 1976, 86, 189281
- I.L. Knuniants, L.S. German, B.A. Diatkin., Izv.. Akad. Nauk. SSSR, Ser. Khim., 1956, n.11, p.1353
- N. Ishikawa, A Takaoka, H. Jwakiry., Bull. Chem. Soc. Jpn.,1979, 52(11), 3377, CA, 1979, 92, 93823
- S. Watanabe, T. Fujita, Y. Usui., J. Fluor. Chem., 1986, 31, p.247
- S. Watanabe, T. Fujita, K. Suga and others., J. Amer. Oil Chemists Soc.,1983, 60, p.1678
- S. Watanabe, T. Fujita, K. Suga and others., Synthesis, 1984, n.1, p.31
- S. Watanabe, T. Fujita, K. Suga and others., J. Amer. Oil Chemists. Soc., 1984, 61, n.9 p.1479
- S. Watanabe, T. Fujita, Y. Usui and others., J. Fluor. Chem., 1986, 31, p.135
- S. Watanabe, T. Fujita, M. Sakamoto and others., J. Fluor. Chem., 1988, 38, n.2, p.243
- S. Watanabe, T. Fujita, M. Sakamoto., J. Fluor. Chem., 1988, 39, n.1, p.17
- T. Kobayashi, M. Maeda, H. Komatsu., Chem. Pharm. Bull., 1982, 30, n.9, p.3082, CA, 1982, 98, 161017
- S. Watanabe, T. Fujita, M. Sakamoto and others., J. Fluor. Chem., 1987, 36, n.3, p.361
- JP Appl., 82 150669, 1981-1982, Daikin Kogyo Co. Ltd., CA, 1982, 98, 53858
- JP Appl., 83 57357, 1981-1983, Daikin Kogyo Co. Ltd., CA, 1983, 99, 121842
- T. Kitazume, N. Ishikawa., Chem. Lett., 1980, 10, p.1327
- J.F. Clark, D.P. Schumaher, Wu Guang Zhong., WO Appl., 92 07824, 1990-1992, Schering Corp., CA, 1992, 117, 111273
- T. Ando, N. Koseki, I. Yasuhara., Biosci Biotechnol Biochem, 1992, 56, n.10, p.1581, CA, 1993, 118, 102252
- K. Ogura and others., Tetrahedron Lett., 1997, 38, n.29, p.5173
Fluorine Notes, 2000, 8, 5-6


